
Abstract
Aim:
To report our methods for expression and purification of α7 nicotinic acetylcholine receptor (α7-nAChR), a ligand-gated pentameric ion channel and an important drug target.
Methods:
α7-nAChRs of 10 different species were cloned into an inducible BacMam vector with an N-terminal tag of a tandem maltose-binding protein (MBP) and a TEV cleavage site. This α7-nAChR fusion receptor was expressed in mammalian HEK293F cells and detected by Western blot. The expression was scaled up to liters. The receptor was purified using amylose resin and size-exclusion chromatography. The quality of the purified receptor was assessed using SDS-PAGE gels, thermal stability analysis, and negative stain electron microscopy (EM). The expression construct was optimized through terminal truncations and site-directed mutagenesis.
Results:
Expression screening revealed that α7-nAChR from Taeniopygia guttata had the highest expression levels. The fusion receptor was expressed mostly on the cell surface, and it could be efficiently purified using one-step amylose affinity chromatography. One to two milligrams of the optimized α7-nAChR expression construct were purified from one liter of cell culture. The purified α7-nAChR samples displayed high thermal stability with a Tm of 60 °C, which was further enhanced by antagonist binding but decreased in the presence of agonist. EM analysis revealed ring-like structures with a central hydrophilic hole, which was consistent with the pentameric assembly of the α7-nAChR channel.
Conclusion:
We have established methods for crystallization scale expression and purification of α7-nAChR, which lays a foundation for high-resolution structural studies using X-ray crystallography or single particle cryo-EM analysis.
Similar content being viewed by others
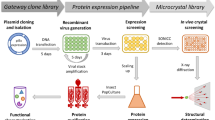


Introduction
Ligand-gated ion channels (LGICs) are among the most important therapeutic targets for various disorders. Approximately 15% of all currently available drugs act through LGICs1. Nicotinic acetylcholine receptors (nAChRs) comprise a major subfamily of LGICs that function at the neuromuscular junction2. nAChRs in the human nervous system have been fine-tuned by evolution to transduce a chemical signal into an electrical signal. nAChRs control the electrical signaling between nerve and muscle cells by opening and closing a gated membrane-spanning pore in response to the neurotransmitter acetylcholine (ACh), which plays a key role in activating the receptor ion channel pore3.
The function of nAChRs is required to maintain appropriate neuronal activities that affect various physiological processes, including sleep, anxiety, and the processing of pain and cognitive functions[4]. nAChR activity is blocked by curare, hexamethonium, and peptide toxins, such as α-bungarotoxin, which is present in the venom of snakes and shellfish5. nAChRs are the molecular targets of nicotine addiction in smokers, and these receptors mediate the positive effects of nicotine on cognition, memory and attention in patients with schizophrenia and neurodegenerative diseases, such as Alzheimer's and Parkinson's diseases6.
nAChRs belong to the superfamily of pentameric ligand-gated ion channels (pLGICs), known as the Cys-loop receptors, which also includes the anion-selective GABAA/GABAC and glycine receptors and cation-selective 5-HT3 serotonin receptors7. nAChRs are some of the most thoroughly studied members of the pLGIC family. Significant efforts in the past 20 years were devoted to structural analyses of nAChRs, which revealed that nAChRs are comprised of five subunits arranged around a central pore that serves as an ion channel. Seventeen nAChR subunits have been identified to date: α1–α10, β1–β4, δ, γ, and ɛ8. Each nAChR subunit is organized into three domains: a large N-terminal extracellular ligand-binding domain (ECD) that contains the characteristic Cys-loop; a transmembrane domain (TMD) consisting of four transmembrane helices (TM1-TM4) and a large cytoplasmic loop that connects TM3 and TM4; and a small C-terminal extracellular domain that follows TM49 (Figure 1). The nAChRs are broadly classified into two subtypes based on their primary sites of expression: muscle-type nicotinic receptors and neuronal-type nicotinic receptors. Muscle-type receptors are composed of α1, β1, γ, and δ subunits in a 2:1:1:1 ratio or α1, β1, δ, and ɛ subunits in a 2:1:1:1 ratio. The neuronal-types are composed of heteromeric combinations of twelve different nicotinic receptor subunits, α2–α10 and β2−β4, for example, (α4)3(β2)2, (α4)2(β2)3, or the homomeric complex (α7)510.

Schematic representation of a nAChR. The topological structure shows that a typical nAChR is composed of three distinct structural and functional entities: a large β-structured extracellular domain (ECD) that comprises the ligand-binding sites, followed by three hydrophobic transmembrane domains (TMD, TM1–TM3), a large intracellular domain (ICD), and a fourth hydrophobic transmembrane domain (TM4). N-linked glycosylation sites are highlighted in red. Schematic view of a nAChR based on the X-ray structure of the 5-HT3A receptor (PDB CODE: 4PIR). A dashed line schematically presents a long fragment of the cytoplasmic loop (shown only for one front subunit) that was not resolved by X-ray structure. The double-MBP tag and TEV protease cleavage site used in this work are schematically represented.
Most of the structural information on nAChRs was obtained for ECD, which contains the orthosteric ligand-binding site. The pentameric structures of acetylcholine-binding protein (AChBP) provide crucial insight into ligand recognition and the ligand-induced conformational changes that trigger channel opening11. The X-ray structure of a chimeric ECD of the human α7-nAChR with AChBP provides an invaluable atomic model for α7 ECD assembly12. Our knowledge of the overall topology of the TMD and intracellular domain (ICD) of nAChRs have largely relied on a 4 Å cryo-EM model of the Torpedo marmarota nAChR13, until recent high-resolution structures of the homologous bacterial pLGICs from Erwinia chrysanthemi (ELIC)14 and Gloebacterviolaceous (GLIC)15 were obtained. The Caenorhabditis elegans glutamate-gated chloride channel (GluCl)16 was the first eukaryotic pLGICs structure solved, and it provided crucial insights into channel gating and ion permeation. Two groups presented the X-ray structures of two mammalian Cys-loop receptors in 2014: the human GABAA receptor17 and the mouse serotonin 5-HT3 receptor18. These two structures offer the newest insights into the assembly and signaling mechanisms of pLGICs and enhance our current understanding of neurotransmission.
The endogenous ligand acetylcholine is a pan agonist that activates all nAChR subtypes. Similarly, many nAChR ligands, such as nicotine and epibatidine, target more than one nAChR subtype19. The lack of specificity of some nicotinic agonists is well known, and it is a potential problem for the treatment of illnesses that require the targeting of a specific nAChR subtype20. The identification of a more subtype-selective nAChR drug that is devoid of side effects is one goal of drug discovery research, and the structure of the full-length nAChR would provide critical insight for the rational design of specific nAChR ligands21. However, structural studies have been hampered by difficulties in obtaining sufficient quantities of highly purified nAChRs for crystallization screening. We report our extensive efforts in the expression and purification of nAChRs. We obtained milligram amounts of purified nAChR protein for crystallization and structural determination using a BacMam expression system in HEK293F cells.
Materials and methods
Construct design and molecular cloning
The original amino acid sequences of α7-nAChRs were aligned using the ClustalW program22. The coding regions of α7-nAChRs from 10 different species were synthesized based on sequence alignment. We designed a series of modified constructs according to the alignment and published structure information. All of the original and modified α7-nAChR cDNAs were cloned into EcoRI/NotI-digested BacMam mammalian cell expression vectors (BacMam expression system, Invitrogen, Cergy Pontoise, France) using conventional molecular cloning technology. An N-terminal double-MBP tag and a TEV cleavage sequence were introduced into the vector to facilitate the expression and purification of the target membrane proteins. The sequences of all clones were confirmed using automated DNA sequencing.
Cell culture and transfection
For routine cell culture, HEK293F cells (FreeStyle293F cells, Invitrogen) were grown as an adherent monolayer in 175 cm2 tissue culture flasks containing 10 mL of high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (Gibco, Invitrogen). The cells were cultured at 37 °C and 95% air/5% CO2 in a humidified incubator. HEK293F cells were transfected using a modified insect cell virus (baculovirus) in accordance with the commercial kit's instruction, and recombinant baculoviruses were obtained following the manufacturer's protocol (BacMam expression system, Invitrogen).
Small-scale expression and construct screening
The HEK293F cells were resuspended and spread onto a 12-well cell culture plate for small-scale expression. The BacMam viruses were transfected into cells at a 1:10 (v/v) ratio. The cells were induced with 1 μg/mL doxycycline (Sigma-Aldrich, St Louis, MO, USA) 2 h later and harvested 18–24 h following induction. The transfected cells were solubilized, and the expression levels of the target protein were detected by Western blot and an anti-MBP monoclonal antibody (Prospec, East Brunswick, NJ, USA). Bacmids with high protein yields were selected for large-scale expression.
Large-scale expression and purification of recombinant α7-nAChRs
Scale-up and harvesting
For large-scale expression, HEK293F cells from 175 cm2 culture flasks were trypsinized and transferred to a 2.8 L shake flask containing 1 L of the serum-free suspension medium CDM4HEK293 (Hyclone, Logan, UT, USA) plus 4 mmol/L Glutamax-1/20 mmol/L HEPES. Typically, 2.8 L shake flask cultures were seeded at 1×106 cells/mL and transduced by recombinant baculoviruses when the cell density reached 4×106 cells/mL. pLGIC expression was induced with 1 μg/mL doxycycline. The cells were collected 24 h after transduction and pelleted from the medium using centrifugation at 2000×g for 20 min. The cell pellets were stored at -80 °C until used.
Solubilization of membrane proteins
Approximately 1 L of the frozen cell pellets were resuspended in 50 mL of lysis buffer, which consisted of 10 mmol/L Tris/HCl (pH 7.4), 10 mmol/L KCl, 10 mmol/L MgCl2 with 5 mmol/L iodoacetamide and a protease inhibitor cocktail (Roche), and incubated in ice for 20 min. The cells were homogenized for 5 min using a dounce homogenizer with a tightly fitting pestle (Thermo Scientific, Rockford, IL, USA). The lysate was centrifuged at 100000×g for 40 min, and the pellets were resuspended in 20 ml solubilization buffer consisting of 20 mmol/L Tris/HCl (pH 7.4), 150 mmol/L NaCl, 1% n-decyl-β-D-maltopyranoside (DDM, Affymetrix, Santa Clara, CA, USA) plus a protease cocktail (Roche, Basel, Switzerland), followed by gentle agitation for 2 h to solubilize the membranes. Insoluble material was removed using centrifugation for 40 min at 100000×g followed by filtration through a 0.45-μm filter.
Affinity purification and size-exclusion chromatography of α7-nAChRs
Solubilized α7-nAChRs were purified using affinity chromatography and a 5 mL amylose column (GE Healthcare). The amylose resin was equilibrated with 5 column volumes of binding buffer consisting of 20 mmol/L Tris/HCl (pH7.4), 150 mmol/L NaCl, 0.05% DDM before loading. Solubilized α7-nAChRs were loaded onto the column and bound overnight using continuous rotation. The amylose resin was washed with 20 column volumes of binding buffer and eluted with 3 column volumes of the same buffer supplemented with 10 mmol/L maltose. Concentrations of the elution fractions containing the purified α7-nAChR were determined using a Bradford assay and an appropriate standard curve. The fusion nAChR protein was cut using a TEV Protease (Sigma) at a ratio of 1:500 (w/w) for 4 h. The protein was concentrated to approximately 5 mg/ml and further purified using a Superose 6 10/300 GL gel filtration column (GE Healthcare) equilibrated in binding buffer. Protein fractions were identified at OD280, and α7-nAChR fractions with the affinity tag removed were pooled and concentrated for subsequent analysis.
Characterization of the α7-nAChR
Fluorescence microscopy
An N-terminal superfolder GFP tag was introduced into the BacMam α7-nAChR vector. HEK293F cells were grown in routine cell culture, and the tagged receptor was expressed by the addition of a BacMam virus at a 1:10 (v/v) ratio and 1 μg/mL doxycycline (Sigma) 18–24 h before measurement. GFP fluorescence images were recorded using a customized, wide-field epifluorescence microscope (Nikon TE2000, Nikon, Canada) with appropriate excitation and emission filters.
SDS gel electrophoresis
Purified α7-nAChRs were analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in a 5% polyacrylamide stacking gel and 10% polyacrylamide separation gel. The proteins in gels were visualized by staining with Coomassie brilliant blue dye.
Western blotting
Western blotting was conducted as described in the kit's manual (Bio-Rad). The transfected cells from a 12-well cell culture plate were harvested and lysed with lysis buffer (50 μL/well) containing 20 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1% DDM plus protease cocktail (Roche). Samples (20 μL) containing total protein were separated using SDS-PAGE and transferred to PVDF membranes, followed by blocking and incubation with an anti-MBP primary antibody (1:1000) at 4 °C overnight. The blots were incubated with a horseradish peroxidase-conjugated secondary antibody (1:2000) at room temperature for 1 h and developed using a Bio-Rad system.
Thermal stability assay
Thermal stability assays were performed as previously described23. N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) dye (Invitrogen) was dissolved in DMSO (Sigma) at 4 mg/mL as a stock solution, which was kept at -80 °C and diluted 1:40 in a dye dilution solution [10 mmol/L buffer, 150 mmol/L NaCl, 0.05% (w/v) DDM] before use. The thermal denaturation assay was performed in a total volume of 200 μL in a quartz fluorometer cuvette (Starna Cells, Inc, Atascadero, CA, USA). The purified receptor (4 μg) was diluted in the appropriate buffer and ligand solution to a final volume of 200 μL. The concentration of ligand used was ten-fold the Ki value. The diluted dye solution (5 μL) was added to the protein solution and incubated for 30 min at 4 °C in the dark. The mixed solution was transferred to a cuvette, and the data were collected using a Cary Eclipse spectrofluorometer (Varian, USA) with a temperature ramping rate of 1 °C/min over a temperature range from 20 °C to 90 °C. The excitation wavelength was 387 nm, and the emission wavelength was 463 nm. The stability data were processed, and melting temperatures (Tm) were obtained using Prism (GraphPad Software, La Jolla, CA, USA).
Electron microscopy
A purified α7-nAChR (0.3 mg/mL) sample (5 μL) was deposited onto a glow-discharged 400 mesh continuous carbon grid (Beijing Zhongjingkeyi Technology, Beijing, China). The sample was stained with 2% uranyl formate and air-dried. The data were recorded on a Tecnai G2 F20 TWIN transmission electron microscope (FEI) equipped with a field-emission gun operated at 200 kV. Images were recorded at 71000×microscope magnification on a 4k×4k Eagle CCD camera with a pixel size of 1.15 Å per pixel. The defocus ranged from -1.0 to -1.5 μm. A total of 1071 particles were boxed out from 81 micrographs using the e2boxer.py program in EMAN2.124. Reference-free 2D image classification was performed using the EMAN1.9 program refine2d.py25.
Results
An efficient mammalian expression system for the human α7-nAChR
We previously tried various bacterial and baculovirus-insect cell expression systems, but all of these systems failed to express human α7-nAChR. Therefore, we turned our attention to mammalian expression systems. The use of Human Embryonic Kidney 293 (HEK293) cells to express full-length human membrane proteins has become increasingly popular because these cells provide a near-native protein folding and lipid environment26. Several mammalian ion channels, including transient receptor potential channel (TRP)27 and the homopentameric 5-HT3 receptor, have been expressed in HEK293 cells, which suggested a potential solution to the poor expression of α7-nAChR28.
We used the BacMam expression system from Invitrogen for mammalian expression because it offers high levels of protein expression29. A codon-optimized cDNA of full-length human α7-nAChR was cloned into the mammalian cell BacMam expression vector, and the baculovirus was generated from insect cells using standard methods (Figure 2A). We introduced the coding region for an N-terminal superfolder GFP tag into the BacMam vector to quickly assess the efficiency of this system (Figure 2B). The adherent monolayer HEK293F cells displayed a uniform and robust expression of α7-nAChR one day after Bacmid transfection, which was detected as high levels of GFP fluorescence signal on the surface of HEK293F cells (Figure 2C).

Recombinant expression of the human α7-nAChR in HEK293F cells. (A) A flow chart of α7-nAChR expression and purification using the BacMam system. (B) Schematic depiction of the N-terminally GFP-tagged expression construct, in which a superfolder GFP (sfGFP)-tag was introduced into the BacMam vector. (C) The surface expression of the superfolder GFP-tagged α7-nAChR in HEK293F cells was demonstrated using fluorescence microscopy.
Design and optimization of the α7-nAChR expression construct
Once the initial expression system for α7-nAChR was established, it was important to screen various expression constructs for expression levels and protein behavior to assess whether these expression constructs were suitable for structural studies. Several variables exert a pronounced effect on expression levels, homogeneity, and the crystallizability of membrane proteins, including affinity tag type and location, codon use, detergent types, truncations, paralogues or orthologues, ligands, and lipids26. Fortunately, many of these variables can be initially assessed using proteins that are expressed in transiently transfected HEK293F cells.
We first screened the expression levels of α7-nAChR from 10 different species selected by phylogenetic analyses of amino acid sequences (Figure 3). The synthetic cDNA constructs encoding the full-length α7-nAChRs were codon-optimized for expression in mammalian cells and cloned into BacMam vectors. We introduced an N-terminal double MBP-tag and a TEV cleavage sequence, instead of an sfGFP-tag, into the vector for membrane protein expression and purification (Figure 4A). The α7-nAChR fusion proteins were expressed in a small-scale in HEK293F cells, and the expression levels were efficiently detected using Western blotting. Among the 10 different species tested, α7-nAChRs from the zebra finch (Taeniopygia guttata) were expressed at the highest levels (Figure 4B).

Sequence alignment of α7 nAChRs from 10 different species. Secondary structures are shown schematically above the sequences, based on the X-ray structure of the 5-HT3A receptor (PDB CODE: 4PIR). Some of the residues important for construct optimization are highlighted by red arrows. NCBI Reference Sequence: H sapiens, NP_000737.1; G gallus, NP_989512.2; T guttata, XP_002196642.1; O cuniculus, XP_002718333.1; A melanoleuca, XP_002929231.1; C familiaris, XP_545813.2; B taurus, NP_776940.1; D rerio, NP_957513.1; T castaneum, ABS86911.1; A gambiae, XP_314691.2.

Design and optimization of the α7-nAChR expression construct. (A) Schematic depiction of the N-terminal double-MBP expression construct. (B–F) Western blot analysis of α7 nAChRs from 10 different species (B), N-terminally truncated α7 nAChR (C), C-terminally truncated α7 nAChR (D), glycosylation of wild-type (N+) and mutated (N–) receptor (E), and the receptor with a GSA-linker replacement of the intracellular loop (F).
We screened the expression of various constructs with N-terminal and C-terminal truncations, removal of N-linked glycosylation sites, deletion of the predicted flexible intracellular loop between TM3 and TM4, and introduction of chimeric domains to further optimize the expression constructs (Figure 4). We designed 5 different lengths of the N-terminal truncation and 8 different lengths of the C-terminal truncation based on sequence alignment. The data showed that any truncation at the N-terminus reduced protein expression (Figure 4C), and most C-terminal truncations increased expression. The construct that included a deletion of 6 amino acids from the C-terminus exhibited the best expression (Figure 4D). The C-terminal 6 amino acids are not conserved among species, and these amino acids are completely missing in some species (Figure 3).
The α7-nAChR contains 3 putative N-linked glycosylation sites, and the receptor seemed to be heavily glycosylated when expressed in HEK293F cells, as indicated from Western blot (Figure 4E) and SDS-PAGE (Figure 5) analyses. Heterogeneity of the glycosylation pattern in the HEK293F-derived protein may compromise crystallization. Therefore, we mutated the N-glycosylation sites (3 per subunit, total of 15 in each homopentamer) to remove the heterogeneity of the carbohydrates. These mutated receptors (N46A, N90A, and N133A) displayed only slightly reduced expression levels with a reduced molecular weight in SDS gels, possibly due to the absence of glycosylation (Figure 4E).

Large-scale expression and purification of the WT α7-nAChR. (A) Size-column purification of the MBP-tagged (before TEV cleavage) Taeniopygia guttata and untagged (after TEV cleavage) α7-nAChR. (B) SDS-PAGE analysis of the elution fractions from the amylose column (lane 1), the sample digested with TEV protease (lane 2), the pooled fractions from size-exclusion chromatography of the TEV-digested sample (lane 3), the double-MBP tag (lane 4), and TEV protease (lane 5).
The α7-nAChR is predicted to have a large flexible intracellular loop of 100–150 amino acids connecting TM3 and TM4 (Figure 2). This intracellular loop is not as conserved as other regions of the receptors, and deletion of the predicted flexible intracellular loop between TM3 and TM4 increases protein expression but does not affect protein function30. Therefore, we introduced a linker region with 4 to 7 copies of the tripeptide Gly-Ser-Ala (GSA) to replace the intracellular loop of TM3 and TM4. The data showed that all of these GSA-linked receptors had increased expression (Figure 4F).
Large-scale expression and affinity purification
We used suspension cell culture for large-scale expression because it allowed mass production of the receptor. The HEK293F cell line expressing the receptor displayed a high growth rate after adaptation to the suspension culture in small volumes, and it reached 2×106 cells/mL with virtually no cell death. One-liter suspension cultures for large-scale expression were induced at 4×106 cells/mL, and the cells were harvested one day later. This technique typically yielded 10 g of biomass and 2 g of isolated membranes that contained up to 6 mg of α7-nAChR fusion protein. We routinely generated 1–2 mg of pure un-tagged α7-nAChR protein using the purification protocol described below.
The detergent DDM is well suited for the extraction of large amounts of the α7-nAChR fusion protein from membranes, and it was used at a concentration of 1% (w/v) for membrane solubilization and 0.05% for subsequent purification steps. The N-terminal double-MBP tag allowed purification of the receptor fusion protein from the membrane extract in a single step with an acceptable degree of purity (Figure 5B). In addition to serving as an affinity tag, the double-MBP tag was also designed to increase protein solubility and improve the yield of purification. Using this construct in a BacMam system, we obtained the pure fusion receptor with a typical yield of 6 mg from one liter of virus-transfected cells, and the MBP tag of the fusion receptor was removed by digestion with the TEV protease (Figure 5B).
We performed size-exclusion chromatography at the last stage of the α7-nAChR purification using a Superose 6 10/300 column, which is capable of separating proteins with sizes up to ten million Daltons, to isolate the pentameric nAChR from non-native aggregated species and the double-MBP tag. We observed a clear peak that corresponded to the fusion pentameric α7-nAChR receptor in the elution fraction from the affinity purification step (Figure 5A). TEV cleavage of the tag changed the elution profile that yielded a peak at approximately 13 mL. This peak corresponds to an apparent molecular mass of approximate 290 kDa based on a calibration of the column (Figure 5A), which corresponded to a pentameric complex of nAChR that is composed of a monomeric subunit with a predicted molecular weight of 58 kDa. The protein peak seemed quite broad (Figure 5), whereas the glycosylation site-mutated receptor appeared to have a sharper peak (Figure 6). The purified nAChR migrated with a mobility that between the 50 and 70 kDa protein markers in SDS-PAGE gels (Figures 5, 6).

Analytical size-exclusion chromatography (SEC) and SDS-PAGE analysis of the optimized α7-nAChR. The Taeniopygia guttata glycosylation site-mutated α7-nAChR (aa26-496) proteins without (26-496/N–) (A, B) and with 7 GSA linker replacement (26-496/N–/7GSA) (C, D) were separated on a Superose 6 10/300 GL gel filtration column (GE Healthcare) at a flow rate of 0.5 mL/min. α7-nAChRs from the pooled fractions after SEC were detected using SDS-PAGE (B, D).
Thermal stability and electron microscopy analysis of the purified receptor
High stability is an important parameter for the crystallization of membrane protein complexes. We used a fluorescence-based thermal stability assay with CPM dye, which dramatically increases its fluorescence intensity upon binding to accessible cysteines, to determine the stability of the purified receptor. Buried cysteine residues were inaccessible to CPM dye in the native state, but these residues become exposed upon thermal denaturation of the protein. The wild-type (WT) receptor and the C-terminally truncated receptors displayed similar melting temperatures (Tm) between 59-60 °C (Figure 7A). The glycosylation site-mutated receptors and the receptor in which the intracellular loop between TM3 and TM4 was replaced with a 7×GSA linker displayed only a slightly decreased Tm of 58 °C (Figure 7A). Notably, inclusion of the antagonists α-bungarotoxin and methyllycaconitine increased thermal stability by 4–5 °C, whereas the agonist epibatidine greatly decreased the thermal stability of the receptor (Figure 7B). The 15 °C difference in Tm between antagonist- and agonist-bound receptors indicates that the antagonist-bound conformation (presumably the closed channel conformation) is more stable than the agonist-bound (presumably open channel) conformation. This result is consistent with the fact that the nAChR channel is only transiently open upon the binding of the physiological agonist acetylcholine.

Thermal stability profiles of the purified α7-nAChR. (A) Proteins from various optimized constructs; (B) Effect of different ligands on the thermal stability of purified α7-nAChRs.
We also performed negative-stain EM analysis of the purified receptor that had a 7×GSA linker between TM3 and TM4. The EM images revealed a wide distribution of aggregating states of the receptor complex with a fraction of single complexes, which displayed a ring-like structure with a central hole (Figure 8). The ring-like structure observed in EM analysis resembled the pentameric structure of the nAChR ECD structure, which is consistent with the assembly of the nAChR channel.

Representative negative-stain EM images of purified α7-nAChRs (26-496/N–/7GSA). Here, α7-nAChR particles are indicated by white boxes, and typical class averages of top and side views are shown in the right panel. The top view image shows a ring-like structure feature with a central hole.
Discussion
The nicotinic acetylcholine receptors are the best-studied members in the pLGIC superfamily. Detailed knowledge of their structures would facilitate the understanding of their functions and the rational design of subtype-specific drugs. However, attempts to solve the crystal structures of these membrane proteins faced many technical challenges. One of the major obstacles was the preparation of nAChR proteins in sufficient quantity and quality. This study reported our extensive efforts in the expression and purification of the α7-nAChR and the biochemical and EM characterization of the purified receptor.
Several attempts were made to express full-length pLGICs from different species in prokaryotic and eukaryotic expression systems. ELIC14 and GLIC15 were expressed in bacteria as fusion proteins with maltose-binding protein, which yielded water-soluble ligand-binding proteins with sizes corresponding to pentamers. Breakthrough studies were also made for GluCl16 and 5-HT3A18, which were expressed in insect cells and HEK293s cells, respectively. Our work demonstrated that α7-nAChRs can be expressed in HEK293F cells using a BacMam expression system and receptor modifications, including C-terminal truncation, replacement of the intracellular loop with GSA linkers, and the removal of glycosylation sites, which improved protein expression and behavior. The purified protein appeared to be quite stable with a Tm approaching 60 °C. The presence of antagonists further increased the thermal stability of the purified receptor, but the presence of agonist decreased the stability, which implies that the open-channel conformation is less stable than the closed conformation. EM analysis of single isolated complexes revealed a ring-like structure that is consistent with the pentameric assembly of the channel.
Our primary goal was to obtain a crystal structure of α7-nAChR. However, we failed to generate crystals of this important protein complex despite our exhaustive efforts in crystallization screening. EM images (Figure 8) indicated that the purified receptor from the final gel filtration fractions contained many forms of micro-aggregation, which may prevent crystallization. The purified receptor appeared to have high thermal stability and respond to the presence of agonist and antagonist, but it was still prone to aggregation that prevented crystallization. There are several ways to overcome this aggregation problem, including protein engineering, detergent and additive screening, and the inclusion of antibody fragments into crystallization. The recent success of nanobody-facilitated crystallization of the 5-HT3A complex is particularly encouraging, and this technique could be similarly applied to α7-nAChRs. The expression and purification methods reported here should pave the way for future high-resolution studies of α7-nAChR by crystallization or single-particle cryo-EM analysis.
Author contribution
H Eric XU and Hao CHENG designed research; Hao CHENG, Si-wei ZHANG and Zhong-shan WU performed research; Yao CONG, Chen FAN and Zhi-cheng CUI contributed electron microscopy analysis; Cheng-hai ZHANG and Yi JIANG contributed BacMam expression system; H Eric XU, Yao CONG and Hao CHENG analyzed data; H Eric XU, Karsten MELCHER and Hao CHENG wrote the paper.
References
Hurst R, Rollema H, Bertrand D . Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther 2013; 137: 22–54.
Miller PS, Smart TG . Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31: 161–74.
Fasoli F, Gotti C . Structure of neuronal nicotinic receptors. Curr Top Behav Neurosci 2015; 23: 1–17.
Paterson D, Nordberg A . Neuronal nicotinic receptors in the human brain. Prog Neurobiol 2000; 61: 75–111.
Role LW, Berg DK . Nicotinic receptors in the development and modulation of CNS synapses. Neuron 1996; 16: 1077–85.
Mehta M, Adem A, Kahlon MS, Sabbagh MN . The nicotinic acetylcholine receptor: smoking and Alzheimer's disease revisited. Front Biosci (Elite Ed) 2012; 4: 169–80.
Corringer PJ, Le Novere N, Changeux JP . Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol 2000; 40: 431–58.
Gotti C, Clementi F . Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 2004; 74: 363–96.
Hogg RC, Raggenbass M, Bertrand D . Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol 2003; 147: 1–46.
Wallace TL, Bertrand D . Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin Ther Targets 2013; 17: 139–55.
Yakel JL . Gating of nicotinic ACh receptors: latest insights into ligand binding and function. J Physiol 2010; 588: 597–602.
Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM, et al. Ligand-binding domain of an alpha7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci 2011; 14: 1253–9.
Unwin N . Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 2005; 346: 967–89.
Hilf RJ, Dutzler R . X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 2008; 452: 375–9.
Hilf RJ, Dutzler R . Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 2009; 457: 115–8.
Hibbs RE, Gouaux E . Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 2011; 474: 54–60.
Miller PS, Aricescu AR . Crystal structure of a human GABAA receptor. Nature 2014; 512: 270–5.
Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, et al. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 2014; 512: 276–81.
Picciotto MR, Higley MJ, Mineur YS . Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 2012; 76: 116–29.
Changeux JP . Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci 2010; 11: 389–401.
Rahman S . Nicotinic receptors as therapeutic targets for drug addictive disorders. CNS Neurol Disord Drug Targets 2013; 12: 633–40.
Thompson JD, Higgins DG, Gibson TJ . CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–80.
Alexandrov AI, Mileni M, Chien EY, Hanson MA, Stevens RC . Microscale fluorescent thermal stability assay for membrane proteins. Structure 2008; 16: 351–9.
Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 2007; 157: 38–46.
Ludtke SJ, Baldwin PR, Chiu W . EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 1999; 128: 82–97.
Chaudhary S, Pak JE, Pedersen BP, Bang LJ, Zhang LB, Ngaw SM, et al. Efficient expression screening of human membrane proteins in transiently transfected human embryonic kidney 293S cells. Methods 2011; 55: 273–80.
Liao M, Cao E, Julius D, Cheng Y . Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013; 504: 107–12.
Hassaine G, Deluz C, Tol MB, Li XD, Graff A, Vogel H, et al. Large scale expression and purification of the mouse 5-HT3 receptor. Biochim Biophys Acta 2013; 1828: 2544–52.
Dukkipati A, Park HH, Waghray D, Fischer S, Garcia KC . BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr Purif 2008; 62: 160–70.
Jansen M, Bali M, Akabas MH . Modular design of Cys-loop ligand-gated ion channels: functional 5-HT3 and GABA rho1 receptors lacking the large cytoplasmic M3M4 loop. J Gen Physiol 2008; 131: 137–46.
Acknowledgements
This work was supported in part by the Jay and Betty Van Andel Foundation, Ministry of Science and Technology (China) grants 2012ZX09301001 and 2012CB910403, 2013CB910600, XDB08020303, 2013ZX09507001, XDB08030201, 13JC1406300, 31222016, 31270771, and Shanghai Science and Technology Committee grant 13ZR1447600, 13JC1406300. We thank Dr Yi SHI for advice on figure preparation and Yue-ming ZHENG and Zhao-bing GAO for consultation on experimental design.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cheng, H., Fan, C., Zhang, Sw. et al. Crystallization scale purification of α7 nicotinic acetylcholine receptor from mammalian cells using a BacMam expression system. Acta Pharmacol Sin 36, 1013–1023 (2015). https://doi.org/10.1038/aps.2015.34
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.34
Keywords
This article is cited by
-
Uncovering the lipidic basis for the preparation of functional nicotinic acetylcholine receptor detergent complexes for structural studies
Scientific Reports (2016)
-
Ion channels gated by acetylcholine and serotonin: structures, biology, and drug discovery
Acta Pharmacologica Sinica (2015)
