
Abstract
MicroRNAs (miRNAs) are small non-coding RNAs functioning as regulators of hematopoiesis. Their differential expression patterns have been linked with various pathological processes originating from hematopoietic stem cells (HSCs). However, limited information is available regarding the role of miRNAs in myelodysplastic syndrome (MDS). Using miRNA arrays, we measured expression of 1,145 miRNAs in CD34+ bone marrow cells obtained from 39 MDS and acute myeloid leukemia (AML) evolved from MDS patients, and compared them with those of six healthy donors. Differential miRNA expression was analyzed and a panel of upregulated (n=13) and downregulated (n=9) miRNAs were found (P<0.001) in MDS/AML patients. An increased expression of a large miRNA cluster mapped within the 14q32 locus was detected. Differences in miRNA expression of MDS subtypes showed a distinction between early and advanced MDS; an apparent dissimilarity was observed between RAEB-1 and RAEB-2 subtypes. In early MDS, we monitored upregulation of proapoptotic miR-34a, which may contribute to the increased apoptosis of HSCs. Patients with 5q deletion were characterized by decreased levels of miR-143* and miR-378 mapped within the commonly deleted region at 5q32. This is an early report describing differential expression in MDS CD34+ cells, likely reflecting their disease-specific regulation.
Similar content being viewed by others


Introduction
Myelodysplastic syndrome (MDS) is a heterogeneous group of clonal preleukemic conditions with high risk of transformation into secondary acute myeloid leukemia with multilineage dysplasia (AML with MLD) occurring in approximately 30–40% of MDS cases. Individuals with MDS exhibit blood cytopenias, ineffective hematopoiesis and usually hypercellular or normocellular bone marrow (BM).1 Progression of the disease is assumed to be a multistep process of acquisition of several successive genetic abnormalities in a normal hematopoietic stem cell (HSC), which ultimately leads to malignant transformation and expansion of the MDS clone.2 According to the World Health Organization3 classification system, MDS is divided into several subtypes with different characteristics and prognoses: 5q− syndrome, refractory anemia (RA), RA with ringed sideroblasts (RARS), refractory cytopenia with MLD (RCMD) and RA with excess blasts (RAEB-1 and RAEB-2).
MicroRNAs (miRNAs) are small (∼21nucleotides) endogenous non-coding RNAs that negatively regulate gene expression of complementary mRNAs. Many crucial cellular processes are under control of miRNAs and their deregulation is implicated in the disease development and/or progression.4 Only a limited number of reports investigating miRNA deregulation in MDS have been published to date.5, 6, 7, 8, 9 Pons et al.5 measured gene expression of 25 mature miRNAs in mononuclear cells (MNCs) isolated from BM and peripheral blood of MDS patients. The authors reported an overexpression of miRNA cluster miR-17-92 in MDS and differential expression of miR-15a and miR-16 between low-risk and high-risk subgroups of patients. Hussein et al.9 measured expression profiles of 365 miRNAs in total BM cells and demonstrated upregulation of the miR-1/miR-133a cluster. These studies examined miRNA expression in unsorted blood cells; however, gene expression profiles remarkably differ between various cell types.10 Thus, increase of the blast count in high-risk MDS influences miRNA levels when measured in whole-blood samples. Therefore, separation of one cell type is essential for accurate gene expression profiling. As MDS originates from HSC, we believe that the testing of CD34+ fraction is of primary interest.
To understand the impact of miRNA deregulation on MDS development and progression, we performed miRNA microarray analysis on purified CD34+ BM cells obtained from patients with MDS and AML evolved from MDS. In addition, we benefited from our previous gene expression profiling study11 and associated miRNA levels with the appropriate gene expression data.
Materials and methods
Patients
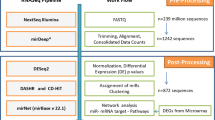
This study comprised 43 BM samples from Czech patients with primary MDS and AML evolved from MDS. According to the WHO 2001 classification criteria, patients’ diagnoses were as follows: 5q− syndrome (n=6), RA (n=4), RARS (n=1), RCMD (n=6), RAEB-1 (n=7), RAEB-2 (n=10) and AML with MLD (n=9). Patients’ age ranged between 31 and 79 years (average 63) and the female/male distribution was 16/27. The clinical characteristics of all patients are summarized in Table 1 and their karyotypes in Supplementary Table 1. BM samples obtained from nine healthy donors were used as controls. Their age ranged between 20 and 40 years (average 30) and the female/male distribution was 3/6. All tested subjects signed the informed consent and the Institutional Review Board approved the study.
CD34+ cell separation
MNCs were separated from BM samples by Ficoll–Hypaque density gradient centrifugation (GE Healthcare, Little Chalfont, UK). CD34+ cells were isolated from MNCs using magnetic cell separation according to the manufacturer's instructions (Direct CD34 Progenitor Cell Isolation MACS Kit, Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of isolated cells (>95%) was tested using FACSAria (Becton Dickinson, San Jose, CA, USA).
RNA extraction
Total RNA was isolated by the method of Chomczynsky and Sacchi12 and subsequently treated with DNase I (Qiagen, Hamburg, Germany). Final RNA integrity was evaluated using Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The RNA integrity number was found to be 9.1 on an average and RNA quality was comparable within the different samples.
miRNA microarray profiling
The human v2 MicroRNA Expression Profiling Kit (Illumina, San Diego, CA, USA) was used for measuring gene expression of 1145 human miRNAs (including 246 novel miRNA candidates denoted by HS codes; annotation file available at http://www.switchtoi.com/annotationfiles.ilmn). Only samples with sufficient RNA yield (39 samples of MDS/AML with MLD and six control samples, input 200 ng of total RNA) were hybridized on Illumina 12-sample BeadArrays. Scanning was performed on the BeadStation 500 instrument (Illumina).
Microarray data analysis
Quality control, background subtraction and quantile normalization of raw data were carried out by GenomeStudio software (Illumina). Only probes that reached detection P-value <0.01 were included for further analyses. Logarithmic transformation of the data was carried out to obtain normal linear distribution. Subsequent analyses were performed using MeV v4.3.2 software.13 The Welch approximate t-test was applied to find out differentially expressed miRNAs between two groups of samples: (i) patients and controls, and (ii) patients with and without del(5q). Analysis of variance (ANOVA) was performed using a 1% false discovery rate threshold to identify differentially expressed miRNAs between different MDS subtypes. Hierarchical clustering of miRNA expression data was carried out using average linkage and Euclidean distance.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The microarray results were confirmed in the complete set of samples (43 samples of MDS/AML with MLD and 9 controls) by qRT-PCR using the RotorGene 3000 apparatus (Corbett Research, Sydney, Australia). qRT-PCR was performed with the following validated TaqMan MicroRNA Expression Assays (Applied Biosystems, Foster City, CA, USA): miR-10a (assay ID 000387), miR-34a (000426), miR-323-3p (002227) and miR-378 (000567). The resulting data were normalized to the endogenous control RNU48 and relative miRNA expression levels were calculated by the ΔΔCT method. Statistical analyses were performed using GraphPad Prism 4 software (GraphPad Software, La Jolla, CA, USA).
miRNA target prediction
Putative miRNA targets were predicted by MicroCosm Targets v.5 (http://www.ebi.ac.uk/enright-srv/microcosm/), TargetScan 5.1 (http://www.targetscan.org/) and PicTar (http://pictar.mdc-berlin.de/) software.
Results
miRNAs differentially expressed between patients and healthy individuals
miRNA expression profiles were measured in CD34+ cells isolated from BM of patients with MDS, AML with MLD and healthy donors (for normalized microarray data, see Supplementary Table 2). Out of 1145 tested miRNAs, 371 miRNAs were not detected and were excluded from further analyses. Subsequently, several statistical tests were applied on the filtered data set to identify differentially expressed miRNAs.
We defined a set of 22 miRNAs differentially expressed between patients and controls (P<0.001): 13 miRNAs were upregulated (miR-299-3p, miR-299-5p, miR-323-3p, miR-329, miR-370, miR-409-3p, miR-431, miR-432, miR-494, miR-654-5p, miR-665, HS_40 and HS_206) and nine miRNAs were downregulated (miR-196a*, miR-423-5p, miR-525-5p, miR-507, miR-583, miR-940, miR-1284, miR-1305 and HS_122.1) in patients (hierarchical cluster in Figure 1). Nine upregulated miRNAs (miR-299-3p, miR-299-5p, miR-323-3p, miR-370, miR-409-3p, miR-431, miR-432, miR-494 and miR-654-5p) mapped within the 14q32 miRNA cluster. In addition, other miRNAs from this cluster displayed a trend of increased expression in ∼80% of MDS/AML patients (data in Supplementary Table 2).

Hierarchical clustering of 22 differentially expressed miRNAs between patients (MDS or AML with MLD) and controls (P<0.001). The binary logarithm of relative miRNA expression changes is expressed by a color gradient intensity scale, as shown at the top. Green color indicates downregulation, red color indicates upregulation of miRNA expression. Each column represents a separate sample and each row a single miRNA. N, control samples; miR*, complementary miRNA variants.
miRNA expression profiles in MDS subtypes
To determine miRNA expression differences between MDS subtypes, we grouped the patients into five categories: 5q− syndrome, low-risk MDS, RAEB-1, RAEB-2 and AML with MLD. Patients with RA (n=1), RARS (n=1) and RCMD (n=6) together formed the ‘low-risk MDS’ group because of a small number of samples. Using the one-way ANOVA test, we identified a set of 45 differentially expressed miRNAs with respect to the disease subtypes. Hierarchical clustering performed for the deregulated miRNAs resulted in the determination of five distinct clusters (Figure 2). miRNAs grouped in the red cluster showed slightly higher expression in low-risk MDS and RAEB-1 than in 5q− syndrome and high-risk patients. On the contrary, miRNAs in the green cluster were downregulated in low-risk MDS and REAB-1 and displayed increased or normal levels in other samples. miRNAs in the blue and brown clusters were downregulated only in the low-risk MDS category. Further, we detected four transcripts (miR-422a, miR-617, HS_63 and HS_305_b; violet cluster) the expression of which correlated with disease progression; their expression was low in normal samples and in patients with early MDS, and high in those with advanced MDS and AML with MLD, with elevation of their expression level between RAEB-1 and RAEB-2.

Hierarchical clustering of 45 differentially expressed miRNAs between different subtypes of the disease (5q− syndrome, low-risk MDS (RA, RARS and RCMD), RAEB-1, RAEB-2 and AML with MLD) (P<0.001). The binary logarithm of relative miRNA expression changes is expressed by a color gradient intensity scale, as shown at the top. Green color indicates downregulation, red color indicates upregulation of miRNA expression. Each column represents a separate sample and each row a single miRNA. Particular miRNA clusters are color-coded on the right side of the heatmap. N, control samples; miR*, complementary miRNA variants.
miRNAs deregulated in patients with 5q deletion
Our MDS subject cohort included six patients with the 5q− syndrome and six patients with advanced MDS bearing the 5q deletion (RAEB-1_3, RAEB-2_3, RAEB-2_4, RAEB-2_9, RAEB-2_10 and AML with MLD_6). A non-parametric Student's t-test was applied to identify miRNAs differentially transcribed in patients with and without deletion in the 5q32 region. Expression levels of 13 miRNAs were elevated and seven of them were reduced in the patients with the deletion (P<0.01, Figure 3).

Differentially expressed miRNAs in patients with and without deletion at the 5q32 region. The binary logarithm of relative miRNA expression changes is expressed by a color gradient intensity scale, as shown at the top. Green color indicates downregulation, red color indicates upregulation of miRNA expression. Each column represents a separate sample and each row a single miRNA. N, control samples; miR*, complementary miRNA variants.
Figure 4 shows expression of miRNAs located in the deleted region in the 5q− syndrome (miR-143, miR-145, miR-146a and miR-378), demonstrating that only expression of miR-378 and miR-143* was significantly reduced in patients with the 5q deletion.

Expression of miRNAs located within the commonly deleted region at 5q32 (miR-143, miR-145, miR-378) or in its proximity (miR-146a) and their complementary variants (miR*) measured by Illumina microarrays. Values are expressed as binary logarithms of fold changes (logFCs). Boxes represent means and 25–75 percentiles and bars indicate the minimum and maximum values for particular patient category compared with the control group (NORM). The 5q− bar represents only patients with pure 5q− syndrome, whereas the bar denoted del(5q) illustrates miRNA expression in all patients with deletion at the 5q32 region (all 5q− together with RAEB-1_3, RAEB-2_3, RAEB-2_4, RAEB-2_9, RAEB-2_10 and AML with MLD_6 patients). *P<0.05, **P <0.01, ***P<0.001; other differences between normal samples and patients with del(5q) were not statistically significant. Expression of miR-145* and miR-146a* was under the detection limit of our experiment.
Validation of microarray data
Using qRT-PCR, expression of four miRNAs was measured in the complete set of samples (43 patients and nine controls): miR-10a, miR-34a, miR-323-3p and miR-378 (Figure 5). We observed differences between miRNA levels in particular disease subtypes comparable to those obtained by microarrays. Moreover, the level of miR-10a inversely associated with progression of the disease (miR-10a level was increased in the 5q− syndrome and RA/RARS samples and reduced in RCMD, advanced MDS and secondary AML, compared with healthy donors).

Validation of microarray data of selected miRNAs using qRT-PCR. Relative fold changes (FCs) of gene expression were measured in the complete set of samples and were calculated by the ΔΔCT method. Values are expressed as binary logarithms of FC. Bars indicate the mean value of logFC plus standard error for particular patient category compared with the control group (NORM). The 5q− bar represents only patients with a pure 5q− syndrome, whereas the bar denoted del(5q) illustrates miRNA expression in all patients with deletion at the 5q32 region (all 5q−, together with RAEB-1_3, RAEB-2_3, RAEB-2_4, RAEB-2_9, RAEB-2_10 and AML with MLD_6 patients).
miRNA targets
To identify target genes of miR-10a, miR-34a, miR-378 and miR-127, we searched within previously published studies and also predicted new putative miRNA targets with potential implication in MDS (Table 2). We attempted to correlate miRNA expression levels with those of their targets measured in our previous study.11 However, we confirmed inverse association of only a small number of target genes: increase of miR-34a corresponded to 70% reduction of SPI1 transcript level in early MDS (P=0.04), reduction of miR-378 to 30% increase of SUFU level in 5q− samples (P=0.01) and miR-127 upregulation to 100% reduction of BCL6 level in all MDS (P=0.02).
Discussion
Microarrays have been anticipated to provide new information to the current knowledge of molecular pathogenesis of MDS. Several studies focused on gene expression signatures of this disorder;11, 21, 22 however, not many crucial questions have been answered. These studies identified many deregulated genes the alteration of which may be critical to the MDS phenotype, but no fundamental implications such as new targets for therapeutic intervention or improvement of prognostic scoring system have been revealed. Recently, our laboratory performed another gene expression profiling of CD34+ progenitors in patients with MDS and secondary AML,11 and the miRNA profiling study presented here continues with further characterization of the same cohort of patients. We expected that the integration of mRNA and miRNA expression data might provide a more complex insight into the disease pathogenesis.
miRNA expression analysis was performed on CD34+ cells obtained from 39 patients (MDS and AML with MLD) and six controls. We identified a set of 22 miRNAs differentially expressed between patients and controls. Interestingly, nine of the upregulated miRNAs were located at 14q32. Upregulation of several miRNAs from this region was also observed in AML bearing t(15;17) translocation.23 14q32 comprises 40 miRNA genes with imprinted expression controlled by a distant differentially methylated region (DMR).24 In our study, we detected significant overexpression of the whole cluster in the majority of MDS/AML patients. We hypothesized that the cluster upregulation might be caused by altered imprinting in DMR, possibly because of uniparental disomy (UPD) and/or deregulated epigenetic mechanisms in 14q32. We read through recent publications whether any defect was detected in this region; however, the absence of copy number variations or UPD found in 14q3225, 26 suggests a change in the methylation or acetylation status or aberrant characteristics of some transcriptional regulators. Limited information is available on the function of 14q32 miRNAs; however, several publications suggested their implication in cell development and oncogenesis. For example, miR-127, a member of the 14q32 miRNA cluster, is involved in B-cell differentiation process through posttranscriptional regulation of BLIMP1 and XBP1.20 In MDS, we detected a negative association of the expression of another miR-127 target, proto-oncogene BCL6,19 functioning also as a transcriptional regulator required for B- and T-lymphocyte terminal differentiation.27 Thus, overexpression of miR-127 may be linked with the downregulation of genes involved in B-cell lineage differentiation previously seen in MDS.11, 28
In CD34+ cells of all MDS subtypes, we confirmed an upregulation of the miR-1/miR-133a cluster (P<0.05, see Supplementary Table 2), which was recently published by Hussein et al.9 In contrast to increased expression in MDS/AML, this miRNA cluster was downregulated in neutrophils from patients with polycythemia vera and essential trombocytosis,29 indicating its different roles in myeloid neoplasms.
5q− syndrome is a MDS subtype characterized by interstitial deletion of the long arm of chromosome 5. The commonly deleted region (CDR) at 5q32 contains 40 genes and several of them may be implicated in hematopoiesis; however, none of the genes seem to be responsible for all characteristics of 5q− syndrome.30, 31, 32 Three miRNAs (miR-143, miR-145 and miR-378) are mapped within the region and miR-146a is located in its proximity. Expression of these miRNAs was examined by Starczynowski et al.33 who showed correlation between del(5q) and the loss of miR-145 and miR-146a in a murine model. However, in human CD34+ and unsorted BM cells, any expression changes of miRNAs encoded in CDR were not detected.7, 9 Our data suggested downregulation of miR-378 and miR-143* (complementary to miR-143 but less abundant), whereas the level of miR-145 and miR-146a remained at the control level. In addition, 50% decrease of miR-378 and miR-143* expression corresponded with the haploinsufficiency status. The discrepancy in the results is probably caused by small numbers of samples in the studies and should be clarified in future.
We hypothesize that an altered epigenetic mechanism at the remaining chromosome, perhaps as a compensational event, may explain why deletion at 5q32 does not lead to reduced expression of the presented miRNAs. Interestingly, we found specific downregulation of miR-143* but not of miR-143 in the 5q− syndrome. Expression of different miRNA variants is usually tissue specific, with lower expression of the complementary variants. It is generally thought that these minor variants are not so important for mRNA regulation. Therefore, 5q deletion might be compensated only in the case of the major variant of miR-143.
In 5q– patients, we observed a significant reduction of the miR-378 level. However, validation of the microarray data revealed its nonspecific deregulation. miR-378 seems to be affected either directly (through chromosome 5 rearrangement as a part of complex karyotype) or indirectly (through deregulation of some upstream target) also in high-risk MDS. This miRNA promotes cell survival, tumor growth and angiogenesis by targeting SUFU gene expression.18 Our previously published gene expression data11 showed an increase of SUFU transcript level in 5q− syndrome patients. Another putative target of miR-378 is transcription factor ETV6. This gene is involved in a large number of chromosomal rearrangements associated with leukemia, including MDS.34, 35 As ETV6 is required for hematopoiesis, it may be a candidate gene generating MDS phenotype. Thus, our findings suggest that the reduction of miR-378 expression may be an event contributing to the pathogenesis of MDS.
Our data provided evidence for miRNA deregulation in particular MDS subtypes. Interestingly, an apparent dissimilarity was observed between RAEB-1 and RAEB-2 expression patterns. The miRNA profile of RAEB-1 patients was more similar to controls and early MDS subtypes, whereas RAEB-2 profiles rather resembled those of secondary AML, suggesting transformation of the disease as early as between RAEB-1 and RAEB-2.
Expression of miR-10a seems to be negatively associated with the progression status of MDS patients. Deregulation of miR-10a in various myeloid disorders has been previously reported (downregulation in AML-unsorted BM cells,23 downregulation in chronic myeloid leukemia CD34+ cells36 or upregulation in BM MNCs of MDS patients5), indicating its different tissue-specific roles in myeloid neoplasms. MiR-10a regulates translation of HOX genes14 that have important roles in regulation of early stages of hematopoiesis, including self-renewal of HSCs.37 Thus, reduction of miR-10a level in advanced MDS may induce (through upregulation of HOX genes) conversion of BM HSCs to leukemic cells, and its decrease might be a marker of MDS progression.
Further, we detected statistically significant overexpression of miR-34a in early MDS (∼10-fold, P<0.001) and its level was also nonsignificantly increased in AML with MLD. MiR-34a is a direct proapoptotic transcriptional target of p5338 and may have, by targeting BCL2,15 a role in the increased apoptosis of BM progenitors seen in low-risk MDS. In addition, we observed an inverse association between expressions of miR-34a and its putative target SPI1 in early MDS. SPI1 encodes transcription factor PU.1, mediating myeloid cell development,39 the expression of which is downregulated in particular subtypes of AML and MDS.40, 41 Taken together, our results suggest that the upregulation of miR-34a in early MDS possibly affects not only apoptosis of HSCs through BCL2 but could also decrease PU.1 expression, which in consequence may impede myeloid signaling and contribute to ineffective hematopoiesis of early MDS.
In this study, we attempted to integrate miRNA expression data with the data from gene expression profiling previously performed in our laboratory.11 However, we confirmed only a limited number of miRNA/mRNA interactions. It is known that miRNAs rather suppress target gene translation, not reflecting its effect on mRNA level. This regulatory impact of miRNAs at the posttranscriptional level may be the reason why mRNA microarrays did not fully meet the expectations of finding new genes eligible for targeted therapy of MDS. We believe that mRNA expression profiling represents preliminary data for future proteomic approaches that will monitor effector molecules, and thus study the real situation in the cell.
This study identified those miRNAs the expression of which is deregulated in all MDS/AML with MLD patients and several miRNAs specific for particular disease subtypes. This is an early report describing complete miRNA expression profiles of MDS CD34+ cells measured by miRNA arrays. Deregulation of several miRNAs detected in this study may contribute to specific features of the disease; however, we are aware that their impact on MDS characteristics has to be fully clarified by functional studies.
References
Aul C, Bowen DT, Yoshida Y : Pathogenesis, etiology and epidemiology of myelodysplastic syndromes. Haematologica 1998; 83: 71–86.
Look AT : Molecular Pathogenesis of MDS. Hematology Am Soc Hematol Educ Program 2005; 2005: 156–160.
Vardiman JW, Harris NL, Brunning RD : The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002; 100: 2292–2302.
Bartel DP : MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297.
Pons A, Nomdedeu B, Navarro A et al: Hematopoiesis-related microRNA expression in myelodysplastic syndromes. Leuk Lymphoma 2009; 50: 1854–1859.
Hussein K, Theophile K, Büsche G et al: Significant inverse correlation of microRNA-150/MYB and microRNA-222/p27 in myelodysplastic syndrome. Leuk Res 2010; 34: 328–334.
Boultwood J, Pellagatti A, Cattan H et al: Gene expression profiling of CD34+ cells in patients with the 5q- syndrome. Br J Haematol 2007; 139: 578–589.
Bousquet M, Quelen C, Rosati R et al: Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J Exp Med 2008; 205: 2499–2506.
Hussein K, Theophile K, Busche G et al: Aberrant microRNA expression pattern in myelodysplastic bone marrow cells. Leuk Res 2010; 34: 1169–1174.
Merkerova M, Belickova M, Bruchova H : Differential expression of microRNAs in hematopoietic cell lineages. Eur J Haematol 2008; 81: 304–310.
Vasikova A, Belickova M, Budinska E, Cermak J : A distinct expression of various gene subsets in CD34+ cells from patiens with early and advanced myelodysplastic syndrome. Leuk Res 2010; 34: 1566–1572.
Chomczynsky P, Sacchi N : Single step Metod of RNA isolation by acid guanidinisothiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156–159.
Saeed AI, Bhagabati NK, Braisted JC et al: TM4 microarray software suite. Methods in Enzymology 2006; 411: 134–193.
Lund AH : miR-10 in development and cancer. Cell Death Differ 2010; 17: 209–214.
Wang X, Liu P, Zhu H et al: miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer's disease, inhibits bcl2 translation. Brain Res Bull 2009; 80: 268–273.
Ichimura A, Ruike Y, Terasawa K, Shimizu K, Tsujimoto G : miR-34a inhibits cell proliferation by repressing MEK1 during megakaryocytic differentiation of K562. Mol Pharmacol 2010; 77: 1016–1024.
Yan D, Zhou X, Chen X et al: MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Invest Ophthalmol Vis Sci 2009; 50: 1559–1565.
Lee DY, Deng Z, Wang CH, Yang BB : MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci USA 2007; 104: 20350–20355.
Saito Y, Liang G, Egger G et al: Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–443.
Leucci E, Onnis A, Cocco M et al: B-cell differentiation in EBV-positive Burkitt lymphoma is impaired at posttranscriptional level by miRNA-altered expression. Int J Cancer 2010; 126: 1316–1326.
Pellagatti A, Cazzola M, Giagounidis A et al: Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010; 24: 756–764.
Miyazato A, Ueno S, Ohmine K et al: Identification of myelodysplastic syndrome-specific genes by DNA microarray analysis with purified hematopoietic stem cell fraction. Blood 2001; 98: 422–427.
Dixon-McIver A, East P, Mein CA et al: Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS One 2008; 3: e2141.
Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaillé J : A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res 2004; 14: 1741–1748.
Gondek LP, Tiu R, O’Keefe CL, Sekeres MA, Theil KS, Maciejewski JP : Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008; 111: 1534–1542.
Heinrichs S, Kulkarni RV, Bueso-Ramos CE et al: Accurate detection of uniparental disomy and microdeletions by SNP array analysis in myelodysplastic syndromes with normal cytogenetics. Leukemia 2009; 23: 1605–1613.
Crotty S, Johnston RJ, Schoenberger SP : Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol 2010; 11: 114–120.
Sternberg A, Killick S, Littlewood T et al: Evidence for reduced B-cell progenitors in early (low-risk) myelodysplastic syndrome. Blood 2005; 106: 2982–2991.
Slezak S, Jin P, Caruccio L et al: Gene and microRNA analysis of neutrophils from patients with polycythemia vera and essential thrombocytosis: down-regulation of micro RNA-1 and -133a. J Transl Med 2009; 7: 39.
Boultwood J, Fidler C, Strickson AJ et al: Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood 2002; 99: 4638–4641.
Lehmann S, O’Kelly J, Raynaud S, Funk SE, Sage EH, Koeffler HP : Common deleted genes in the 5q- syndrome: thrombocytopenia and reduced erythroid colony formation in SPARC null mice. Leukemia 2007; 21: 1931–1936.
Ebert BL, Pretz J, Bosco J et al: Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008; 451: 335–339.
Starczynowski DT, Kuchenbauer F, Argiropoulos B et al: Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med 2010; 16: 49–58.
Mauvieux L, Helias C, Perrusson N et al: ETV6 (TEL) gene amplification in a myelodysplastic syndrome with excess of blasts. Leukemia 2004; 18: 1436–1438.
Odero MD, Vizmanos JL, Román JP et al: A novel gene, MDS2, is fused to ETV6/TEL in a t(1;12)(p36.1;p13) in a patient with myelodysplastic syndrome. Genes Chromosomes Cancer 2002; 35: 11–19.
Agirre X, Jiménez-Velasco A, San José-Enériz E et al: Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res 2008; 6: 1830–1840.
Abramovich C, Humphries RK : Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol 2005; 12: 210–216.
Hermeking H : p53 enters the microRNA world. Cancer Cell 2007; 12: 414–418.
Nagamura-Inoue T, Tamura T, Ozato K : Transcription factors that regulate growth and differentiation of myeloid cells. Int Rev Immunol 2001; 20: 83–105.
D’Alò F, Di Ruscio A, Guidi F et al: PU.1 and CEBPA expression in acute myeloid leukemia. Leuk Res 2008; 32: 1448–1453.
Huh HJ, Chae SL, Lee M et al: CD34, RAB20, PU.1 and GFI1 mRNA expression in myelodysplastic syndrome. Int J Lab Hematol 2009; 31: 344–351.
Acknowledgements
The authors thank Prof Kyra Michalova (Center of Oncocytogenetics, Faculty Hospital and First Faculty of Medicine, Charles University, Prague) for the cytogenetic data. This study was supported by GA CR 301/09/P579 and research task No. 00023736 from the Ministry of Health of the Czech Republic.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Dostalova Merkerova, M., Krejcik, Z., Votavova, H. et al. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. Eur J Hum Genet 19, 313–319 (2011). https://doi.org/10.1038/ejhg.2010.209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.209
Keywords
This article is cited by
-
MicroRNAs as prognostic biomarker and relapse indicator in leukemia
Clinical and Translational Oncology (2017)
-
Transfer RNA detection by small RNA deep sequencing and disease association with myelodysplastic syndromes
BMC Genomics (2015)
-
Myelodysplastic syndromes with 5q deletion: pathophysiology and role of lenalidomide
Annals of Hematology (2014)
-
Expressional changes of genes and miRNA in common megakaryocyte-erythroid progenitors from lower-risk myelodysplastic syndrome
International Journal of Hematology (2014)
-
Aberrant expression of microRNAs in bladder cancer
Nature Reviews Urology (2013)
