
Abstract
5-Hydroxymethylcytosine (5hmC) is a recently characterized epigenetic mark that is particularly abundant in brain tissue and that regulates gene transcription. We have recently begun to understand the important role of 5hmC in brain development, plasticity and disease, but there are currently little data on 5hmC alterations in psychiatric illnesses. Here we report what we believe to be the first genome-wide analysis of 5hmC in the depressed brain. Using AbaSI sequencing, we investigated 5hmC in the prefrontal cortex of depressed (N=19) and psychiatrically healthy controls (N=19). Consistent with previous global 5hmC analyses in other phenotypes, and likely owing to the inter-individual variability in 5hmC content, the distribution of 5hmC across chromosomes and genomic features was not different between groups. We did, however, find 550 CpGs with suggestive evidence of differential hydroxymethylation. Of these, we validated CpGs in the gene body of myosin XVI (MYO16) and insulin-degrading enzyme using targeted oxidative bisulfite sequencing. Furthermore, the enrichment of 5hmC was also associated with changes in the expression of these two genes in depressed suicides. Together, our results present a novel mechanism linking increased 5hmC to depression and provide a framework for future research in this field.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) is a prevalent and debilitating disorder that affects >350 million people worldwide.1 Previous research into MDD has focused on a range of neurobiological features, including cell signaling pathways, neurotransmitters and their receptors, stress response systems, and genetics.2 Most recently, epigenetic mechanisms, which are a collection of chemical and physical changes to the genome that regulate transcription, have been investigated as a possible mechanism to explain the development of persistently depressed mood and associated psychopathology in depressive conditions.3, 4, 5, 6 There are a number of different epigenetic mechanisms and, to date, 5-methylcytosine (5mC) has been the most widely studied epigenetic modification in association with neuropsychiatric conditions.7 Recently, however, it was demonstrated that 5mC is oxidized to 5-hydroxymethylcytosine (5hmC) by the ten–eleven translocation enzymes,8, 9, 10 and that 5hmC is particularly abundant in brain tissue than elsewhere in the organism11, 12, 13 and is positively associated with gene transcription.14, 15, 16, 17 Moreover, 5hmC associates with regulatory genomic regions of genes that are developmentally activated in the human brain. So far, there has been little work investigating 5hmC in psychiatric conditions; however, given the interest of 5hmC for brain disorders, recent studies have begun to explore a potential role of 5hmC in neurodegenerative disorders such as Alzheimer’s disease and Huntington’s disease.18, 19 At present, no common features have been observed in these disorders, suggesting a high diversity of 5hmC genome wide.20 Furthermore, 5hmC in human brain and peripheral tissues shows strong inter-individual variation, thus reducing the power of 5hmC studies compared to 5mC.21, 22
Multiple new methods have been developed to study 5hmC, as bisulfite conversion alone is not able to distinguish between 5mC and 5hmC. Oxidative bisulfite sequencing (oxBS-Seq)23 and ten–eleven translocation-assisted bisulfite sequencing24 provide single-base resolution analyses of both 5hmC and 5mC, but are quite costly on a genome-wide scale. Enrichment-based20 methods, such as 5hmC immunoprecipitation or selective chemical labelling,25 allow for the genome-wide interrogation of 5hmC at a more reasonable cost, but do not provide single-base resolution. In this study, we used a restriction enzyme-based methodology combined with next-generation sequencing, AbaSI sequencing, to present a comprehensive, genome-wide analysis of 5hmC at base resolution in the prefrontal cortex (PFC) of individuals who were affected with major depression at the moment of death and compared them to psychiatrically healthy controls.
This study, which we believe is the first of its kind, adds to the current hypothesis that 5hmC is functionally relevant in brain-related disorders and provides a framework for future studies investigating 5mC and 5hmC in MDD and suicide. Specifically, in characterizing the genomic locations of differentially hydroxymethylated regions, we shed light on an alternative molecular mechanism that may be involved in the development of MDD.
Materials and methods
Subjects
Post-mortem human brain tissue from the PFC, specifically the inferior frontal gyrus, of 19 depressed individuals who died by suicide and 19 psychiatrically healthy controls who died by natural or accidental causes was obtained from the Douglas-Bell Canada Brain Bank. Previous studies have implicated epigenetic mechanisms in the etiology of neuropsychiatric disorders in the PFC,4, 26, 27 as well as other prefrontal cortical regions.28, 29, 30, 31, 32 The tissue was dissected at 4 °C, snap-frozen in liquid nitrogen and stored at −80 °C following standard procedures. The Quebec Coroner’s office assessed the cause of death for each subject, and, subsequently, we obtained information on the subjects’ mental health using psychological autopsies using the Structured Clinical Interviews for DSM-IV axis 1.33 In addition, brain tissue samples from all subjects were assessed for the absence of pathological processes by a neuropathologist. All subjects were French-Canadian males and additional demographic information can be found in Table 1. Age, pH, PMI and RNA integrity number were not significantly different between groups (Table 1). Written informed consent was obtained from next of kin for all subjects, and the Douglas Institute Research Ethics Board approved this study.
AbaSI sequencing
Genomic DNA was extracted from the PFC using QIAGEN’s QIAamp DNA Mini Kit (Qiagen, Toronto, ON, Canada, cat. #51304). Purity and concentrations of genomic DNA were assessed using the Thermo Scientific Nanodrop 1000 spectrophotometer (Carlsbad, CA, USA) and each sample had a 260/280 ratio >1.8. AbaSI sequencing library construction was performed as described previously.14, 34 Briefly, DNA was glucosylated by using UDP-glucose and T4-β-glucosyltransferase (New England Biolabs, Ipswich, MA, USA, cat. #M0357L). The DNA was then digested using AbaSI (New England Biolabs, cat. #R0665S) and custom-biotinylated adaptors were ligated to DNA ends. The DNA was then sheared using the Covaris S220 Focused-Ultrasonicator (Woburn, MA, USA; peak incident power=140; duty %=10; cycles per burst=200; time=120 s). Sheared DNA was captured using Dynabeads MyOne Streptavidin C1 beads (Life Technologies, Carlsbad, CA, USA, cat. #65001). NEBNext End Repair Module (New England Biolabs, cat. #E6050L) and NEBNext dA-Tailing Module (New England Biolabs, cat. #E6053L) were used to generate blunt-ended fragments and create an adenosine tail, respectively. A second set of custom adaptors were ligated to the DNA, followed by PCR amplification using NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs, cat. #E6013AA). Illumina (San Diego, CA, USA) TruSeq indices 1–12 were used to barcode samples. Fifty cycle, single-read sequencing was performed on Illumina’s HiSeq 2000. A minimum of 60 000 000 sequencing reads were obtained per subject. Sequencing quality control metrics can be found in Supplementary Table S1. Importantly, no correlation existed between the total number of 5hmC sites identified in each subject and the respective number of reads, post-mortem interval, age or pH (Supplementary Figures S1a–d).
Data processing
FASTQ reads were initially filtered based on a quality score >30 and were then aligned to the human reference genome (UCSC Hg19) using Bowtie 2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), with the preset parameters of very-sensitive (-D 20 -R 3 -N 0 -L 20 -i S,1,0.50). Duplicates were removed using Picard (http://broadinstitute.github.io/picard/) and realignment was performed using the GATK alignment procedures from the Broad Institute (https://www.broadinstitute.org/gatk/). Subsequent filtering of aligned reads was based on a quality score of ⩾10. Mapped reads with a Q score<10 were discards. Reads with Q>10 that mapped to multiple locations were discarded if they could not be confidently assigned to a single region. Potential 5hmC sites were determined using a custom PERL script34 that identifies cytosine bases at 11–13 bp upstream of the 3′-adaptor sequence, which represents the AbaSI enzymatic cleavage site. 5hmC sites in a CG context were evaluated on a binary scale, based on a present or absent principle. Only those 5hmCGs present in half the population, a level of stringency used to account for inter-individual variability, were used during downstream analyses. Exact specifications are found below.
Density plots
Chromosomes
All 5hmC sites present in at least 19 of the 38 subjects were used to assess chromosomal 5hmC density. For each subject, density was defined as the total number of 5hmC sites present in the subject per chromosome, corrected for the length of the chromosome and the total number of CGs on the chromosome.
Genetic features
5hmC sites present in at least 19 of the 38 subjects were plotted against genomic regions. Similar to the chromosomes, for each subject, density was defined as the total number of 5hmC sites present in the subject per genomic regions, corrected for the length of the region and the number of CGs within the region. All genomic features were defined based on the GRCh37/hg19 genomic annotation downloaded from the UCSC database. Different genic elements, including transcription start sites (TSSs), exons, introns and transcription terminal sites, were defined based on the Ensembl (release 75). As genes can have multiple transcripts, we selected the 5′-most TSS on the positive strand as the single TSS associated with each gene. The reverse (3′-most TSS) was done for genes on the negative strand. We limited downstream analysis to protein-coding genes, resulting in 20 745 TSSs in total. Similarly, annotations for retro-elements (that is, long interspersed nuclear elements and short interspersed nuclear elements) and CpG islands were acquired from the UCSC database. CpG shores were defined as the 2 kb flanking a CpG island. Coordinates of predicted of promoter and enhancer regions were obtained from recently published genome-wide maps of chromatin states in the adult brain midfrontal lobe,35 including H3K4me3, H3K4me1 and H3K27ac. Two types of enhancers were distinguished: active enhancers that were simultaneously marked by distal H3K4me1 and H3K27ac, and poised enhancers that were solely marked by distal H3K4me1.17, 36
Validation by targeted oxidative bisulfite sequencing
Differentially 5hmC analyses and target selection
Because of the binary nature of the data, we performed χ2 analyses to identify differences in hydroxymethylation between control (n=19) and depressed groups (n=19). For each CpG site, we asked whether there is a significant difference in the proportion of samples with a 5hmC mark in the two groups. A P<0.001 was used to define differentially hydroxymethylated sites. Only CpG sites that were hydroxymethylated in at least half of the total number of samples were included in the analysis.
DNA extraction
Genomic DNA was extracted from brain tissue using QIAGEN’s QIAmp DNA Mini Kit (Qiagen, cat. #51304). NanoDrop 2000 spectrophotometer and Quant-IT PicoGreen (Thermo Scientific, cat. #P7589) were used to assess the DNA purity and concentration.
Oxidative bisulfite conversion
The oxidative bisulfite conversion reaction was done using the CEGX True Methyl kit (Cambridge Epigenetix, Cambridge, UK, Cat. #CEGXTMA). Briefly, 1 μg of DNA from all 38 samples was purified and denatured. DNA from each subject was then split in two equal reactions, one of which underwent chemical oxidation followed by bisulfite conversion, and the other underwent mock oxidation (oxidant replaced by water) followed by bisulfite conversion. All bisulfite reactions were cleaned-up using a bead-based purification and final elution was in 30 μl of water. Digestion and sequencing spike-in controls were interrogated pre- and post-sequencing, respectively (Supplementary Figure S2).
Primer design
Using MethylPrimer Express, two pairs of bisulfite sequencing primers were designed for each of the targets to validate. Primer sequences can be found in Supplementary Table S2. For each primer designed, an additional primer was generated containing a universal primer sequence on the 5′ ends of the forward and reverse primers. These universal primers allowed for the subsequent addition of Illumina sequencing primers and indices.
Library preparation
An initial PCR reaction was performed on the bisulfite converted DNA using the Kapa HIFI Uracil+mastermix (Mississauga, ON, Canada) and bisulfite primers without thuniversal primer sequence. The PCR reaction was 95 °C for 3 min, then 40 cycles of 98 °C for 20 s, Tm for 15 s, 72 °C for 15 s, and finished with 72 °C for 1 min. All samples for a given target were then pooled for two rounds of AMPURE purification at a 1 × concentration. A second PCR of only 10 cycles using the same conditions described above was then performed on the purified samples, this time using the primers with the universal primer sequence. The PCR reaction was prufied again with AMPURE at a 1 × concentration. Finally, another 10 cycles of PCR were performed add the Illumina sequencing primers and indices. Two final purifications of 1 × AMPURE were performed to completed the library preparation. Libraires were quantified using the Agilent 2200 TapeStation Instrument (Santa Clara, CA, USA) and DNA 1000 reagents. All samples were normalized based on their molarity and pooled. Samples were sequenced on the Illumina MiSeq sequencer using the V3, 600 cycle kit to perform a 300 bp paired-end sequencing run.
Interrogation of CpGs in target amplicons
Adaptor sequences were trimmed and sequencing reads with a Phred quality score <30 were discarded. The remaining reads were aligned to target regions using Bismark. Regions with <100 × coverage in both the the oxBS and bisulfite-only fractions were removed. Finally, the unconverted CpG percentage was calculated for each CpG site as unconverted read counts divided by total read counts. 5hmC percentage at each CpG site was derived as unconverted CpG ratio in bisulfite-only reaction subtracted by the counterpart in oxidation+bisulfite reaction of the same DNA sample.
Quantitative real-time PCR
RNA extraction
RNA was extracted from PFC tissue using QIAGEN’s miRNeasy mini kit (Qiagen, cat. #217004). The Agilent 2200 TapeStation Instrument was used to determine the concentrations and RNA integrity numbers. Only samples with a RNA integrity number⩾5.0 were used to synthesize complementary DNA.
Quantitative real-time PCR settings
Unless otherwise stated, all qRT-PCRs were performed using 5 ng of complementary DNA in a 10 μl total reaction volume. Reactions were run on Applied Biosystems’ (Carlsbad, CA, USA) QuantStudio 6 Flex System, using a comparative Ct approach (relative quantification) with SYBR Green reagents. The termocycler protocol was: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s, then 60 °C for 1 min. Analyses were performed using Applied Biosystems’ QuantStudio Real-Time PCR Software v1.1.
Endogenous control validation
To determine which endogenous control would be suitable for our analyses, we performed quantitative real-time PCR using primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), beta-actin (ACTB), importin 8 (IPO8) and RNA polymerase II subunit A (POLR2A) in multiple brain regions from control subjects. Analyses showed that ACTB, IPO8 and POLR2A showed variability between brain regions, whereas GAPDH showed both the strongest stability across brain regions and the smallest variation between subjects (Supplementary Figure S3). As a result, GAPDH was chosen as the endogenous control for downstream analyses, which is in line with previous work using similar samples.26, 27 Primer sequences for the endogenous controls and targets are list in Supplementary Table S3.
Statistical analyses
Statistical analyses were primarily performed using GraphPad Prism 6 (La Jolla, CA, USA). Grubb’s outlier analysis was performed before any other statistical computation, as well as a Shapiro–Wilk normality test. Two-way analysis of varinace was used for 5hmC density analyses across chromosomes and genomic features. χ2 analysis was performed using R. Unless otherwise mentioned, statistical significance was set at P<0.05. Error bars=s.e.m.
Results
No differences in 5hmC density across chromosomes and genomic features
In this study, we utilized AbaSI sequencing, a restriction enzyme-based approach that has been successfully used in both embryonic stem cells34 and post-mortem brain,14 to evaluate 5hmC levels in the PFC of depressed individuals (N=19) as compared to psychiatrically healthy controls (N=19). Neither the total (controls, n=17 368 538; depressed, n=17 367 367) nor the mean (controls, n=4 277 308±319 846; depressed, n=4 163 916±280 867; P=0.79) number of 5hmC sites identified across the depressed suicides differed from those of the psychiatrically healthy controls (Figures 1a and b). Similarly, both within and between groups, 5hmC sites showed comparable overlap from one subject to the next (Figure 1c). Furthermore, no significant interaction was observed between controls and MDD for 5hmC density across chromosomes (F(23,828)=0.2532, P=0.99; Figure 1d) and, as has been reported before, in human brain.14, 15 There was, however, a significant main effect of chromosome (F(23,828)=127.7, P<0.0001), suggesting that 5hmC densities differ between chromosomes, regardless of phenotype. Similarly, no significant interaction was observed between controls and MDD for 5hmC density across genomic features (F(8,288)=0.02445, P>0.99); however, a significant main effect of genomic feature was present (F(8,288)=918.6, P<0.0001) (Figure 1e). Specifically, we observed an increase in 5hmC density in the exons and enhancer elements, although being depleted in CpG islands and repetitive elements.

Genome-wide similarities in 5hmC density between depressed individuals and controls. (a) The total number of unique 5hmC sites identified in depressed individuals (n=17 367 367) was similar to that observed in controls (n=17 368 538). (b) Similarly, no differences exist in the mean number of unique 5hmC sites identified per subject per group (controls, n=19, mean=4 277 308±319 846; depressed, n=19, mean=4 163 916±280 867). (c) Pairwise comparisons on the percent of overlapping 5hmC sites identified between any two subjects were comparable, regardless of group. Two-way analysis of varinaces showed that 5hmC density was highest in exons and enhancer regions; however, 5hmC density between cases and controls was equivalent across (d) all chromosomes (F(23,828)=0.2532, P=0.99) and (e) genomic features (F(8,288)=0.02445, P>0.99). Error bars=s.e.m. 5hmC, 5-hydroxymethylcytosine; MDD, major depressive disorder; MYO16, myosin XVI; TSS, transcription start site.
Site-specific differential hydroxymethylation in the depressed brain
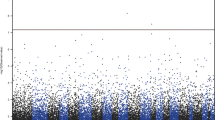
Considering the high level of inter-individual variability in 5hmC (Figure 1c), an observation that has been consistently reported in previous 5hmC brain studies,14, 22 we aimed to focus our analyses on 5hmC sites that are likely to account for a substantial proportion of the total phenotypic variability. To do so, we developed three levels of stringency: low, intermediate and high, which represented all 5hmC sites common to ~25%, 50% and 75% of the total sample, respectively. As the total number of 5hmC sites in the intermediate stringency (n=2 048 613) was closest to the mean number of 5hmC sites observed per subject regardless of group (n=4 220 612±210 144), we chose to look for site-specific differential hydroxymethylation on all 5hmC sites present in at least half of the samples (Figure 2a). Mean coverage of these 2 048 613 sites was 2.97 × and 3.13 × in controls and MDD, respectively. Although no individual site was significant once genome-wide correction was applied, site-specific 5hmC analyses identified 550 suggestive differentially hydroxymethylated sites according to established statistical criteria for genome-wide studies (P<0.001),37 of which 165 5hmC sites were enriched and 385 were depleted in depressed brains (Supplementary Tables 4 and 5). Of these, we chose to validate CpG sites with increased likelihood of being both functionally and phenotypically relevant. To do so, because of the association of gene-body 5hmC and gene expression,14, 15, 16, 17 we narrowed our list to CpG sites present in the body of brain-expressed genes previously shown to be associated with neurodevelopmental or neuropsychiatric diseases or disorders of interest (Table 2).

Site-specific changes in 5hmC content at the MYO16 and IDE loci. (a) Using an intermediate stringency, that is, 2 048 613 5hmC sites common to at least 50% of the population (n=19, outlined by red square), we observe 550 cytosine with differential 5hmC content between depressed individuals and controls (P<0.001). Of these, oxBS-Seq was used to validate an enrichment of 5hmC in the gene bodies of (b) MYO16 and (c) IDE loci (P<0.05). (d, e) Furthermore, at these loci, we observe an inverse relationship between 5mC and 5hmC content (MYO16: r2=0.5720, P<0.0001; IDE: r2=0.4189, P<0.0001). Error bars=s.e.m. *P<0.05; #P⩽0.1. 5hmC, 5-hydroxymethylcytosine; IDE, insulin-degrading enzyme; MYO16, myosin XVI; oxBS-Seq, oxidative bisulfite sequencing.
To validate suggestive differential 5hmC sites of interest, we processed DNA extracted from the PFC of the same 38 subjects (n=19 per group) using a different protocol, namely, oxBS, followed by target amplicon sequencing. Analyses were performed on all CpGs within the target amplicon. oxBS confirmed the increase in 5hmC in depressed brains in myosin XVI (MYO16) at position 109 344 927 (P=0.033) and a statistical trend towards increased 5hmC at the neighboring CpG position 109 344 932 (P=0.10; Figure 2b). There was also an increase in 5hmC in two of the three neighboring CpGs in the insulin-degrading enzyme (IDE) gene (P=0.081 and P=0.0054, respectively), as well as in the mean 5hmC levels across the target region (P=0.025; Figure 2c). As we used oxBS to validate our results, we obtained 5mC, in addition to 5hmC levels in the regions investigated. Although there were no changes in 5mC levels across the targeted CpGs in these two genes (Supplementary Figure S4), there was a strong negative correlation between 5mC and 5hmC in both MYO16 (r2=0.5720, P<0.0001) and IDE (r2=0.4189, P<0.0001; Figures 2d and e).
Gene expression changes in MYO16 and IDE
To determine whether the observed increases in 5hmC in the gene bodies of MYO16 and IDE were associated with changes in gene expression, RNA was extracted from the PFC of the same depressed individuals and psychiatrically healthy controls and gene expression was analyzed by quantitative real-time PCR. For MYO16, we observed an increase in gene expression in depressed suicides (P=0.0020), which was in the expected direction (Figure 3a). However, for IDE, we observed a decrease in gene expression in depressed suicides (P=0.017; Figure 3b). Importantly, as per the Wash U Epigenome Browser (http://epigenomegateway.wustl.edu/), the genomic landscape surrounding these differential 5hmC sites are quite different. Those of MYO16 are located in a quiescent and open chromatin region, whereas those of IDE are found in a region of weak transcription within a long interspersed nuclear elements.

MYO16 and IDE gene expression changes in depressed individuals. (a) MYO16 overexpression is observed in depressed individuals (RQ mean=1.415±0.1134) compared to controls (RQ mean=0.9756±0.08981), whereas that of (b) IDE is decreased in depressed individuals (RQ mean=0.8021±0.07637) compared to controls (RQ mean=1.262±0.1566). *P<0.05. Error bars=s.e.m. IDE, insulin-degrading enzyme; MYO16, myosin XVI; RQ, relative quantification.
Discussion
In this study, we analyzed global and site-specific 5hmC in the PFC of depressed suicides using AbaSI sequencing, a high-throughput restriction enzyme-based technique. Although our results show no global difference in 5hmC density across chromosomes or genomic features, we do observe 550 CpG sites that show suggestive differential hydroxymethylation in depressed suicides compared to psychiatrically healthy controls. Among those, increased 5hmC in the gene bodies of MYO16 and IDE were validated using targeted oxBS-Seq, and both genes showed dysregulated expression in depressed suicides.
MYO16, located on chromosome 13, has been previously implicated in brain development,38 due to its association with the phosphoinositide 3-kinase signaling pathway.39 Genomic variants within MYO16 and increased MYO16 gene expression in patients with schizophrenia40 have also linked this gene to neuropsychiatric disorders. Of particular relevance to this study, phosphoinositide 3-kinase signaling includes the dirsupted-in-schizophrenia locus, which has been shown to be a risk factor in MDD.41
In this study, we too show an increase in MYO16 gene expression, albeit in depressed suicides, that is associated with increased MYO16 gene-body 5hmC. Of interest, the observed differential 5hmC sites are located in a putative quiescent region of open chromatin. Not only is gene-body 5hmC associated with active transcription, but 5hmC has also been shown to be more abundant in both active and poised enhancers.14, 17 The presence of 5hmC in these regions could indicate a transition from 5mC to 5hmC in an effort to recruit histone marks or other transcription factors to active gene transcription. Future studies should better characterize whether this may represent a feature of the etiology of depressive disorders.
IDE is a gene encoding the IDE known to be expressed in insulin-responsive tissues.42 The increase in 5hmC in the gene body of IDE and the accompanying decrease in IDE gene expression seen in depressed suicides were unexpected, given previous reports showing a positive correlation between gene-body 5hmC and gene expression.14, 15, 16, 17 However, this correlation is not as firm in lowly expressed genes, such as IDE.14, 16 Furthermore, the enrichement of 5hmC has also been observed in the extended promoter regions of repressed genes,43 where it has been shown to negatively influence gene expression in psychiatric phenotypes.44, 45
IDE downregulation has been implicated in the development of neurodegenerative disorders, specifically Alzheimer’s disease.46, 47 In the context of depressive disorders, one hypothesis is that decreased expression of IDE in the brain may lead to increased levels of insulin, which elevates levels of reactive oxygen species and strengthens inhibitory GABAergic synapses.48 Interestingly, the accumulation of reactive oxygen species has been implicated in a number of psychiatric disorders49, 50 and it has been suggested that oxidative stress is a potential biomarker of MDD.51 Furthermore, previous expression microarray data from our lab in other cortical brain regions suggest that genes related to this hypothesized insulin-dysregulated pathway may also display differences in individuals with MDD who died by suicide (unpublished data). Taken together, although the association between increased 5hmC and decreased IDE expression is opposite to the expected positive correlation, it is possible that increased activation of the insulin-signalling pathway may result in increased oxidative stress, thereby contributing to the risk for MDD.
The seemingly opposing functions of 5hmC in regulating MYO16 and IDE gene expression are likely explained by the genomic location of the dysregulated 5hmC. It is widely accepted that multiple epigenetic modifications may act together to alter gene transcription. In this particular case, the enriched 5hmC locus in MYO16 is located in an open chromatin region and is, therefore, accessible by other chromatin marks. On the other hand, the enriched 5hmC locus in IDE sits in a region of weak transcription, which can explain the inverse association between 5hmC levels and gene expression. In its entirety, our results add to the notion that differences in genomic landscapes likely infer functional variability of epigenetic marks, thereby explaining the opposite directions of the gene expression differences in these genes in depressed suicides.
This manuscript adds support for the association of both IDE and MYO16 in the etiology of psychiatric disorders. Furthermore, our results present insights into the potential functional role of 5hmC in the depressed brain. Importantly, regulation of gene expression is highly complex and may involve chromatin modifications and small RNAs, in addition to cytosine methylation and hydroxymethylation, each of which may work together to regulate gene expression through multiple mechanisms. The results presented here discuss the association of several CpGs located at two loci of relatively large genes. As such, it would be unjustified to speculate that this 5hmC locus is the main regulator of this gene. Rather, these loci may represent a target for regulatory machinery, in addition to other proximal or distal regulatory regions. Similarly, MDD and suicide are complex and heterogeneous phenotypes that are thought to be determined by a variety of predisposing and precipitating factors. It is more appropriate to think that these changes in the epigenetic landscapes and the expression of these genes are more likely to be involved in a cascade of neurobiological events leading to the etiology of depressive disorders. As such, these genes likely represent a piece of a much larger puzzle that defines the neurobiological alterations that exist in these depressed individuals. Nevertheless, taken together, these data add to the incipient literature, suggesting that 5hmC is involved in brain-related processes and pathological conditions.
This study, however, is not without limitations. As it is always the case in human studies, there are many sources of inter-individual variability between subjects and groups. Although we made an effort to control for common technical and clinical confounders, it is not possible to control for all possible covariates. In terms of the data, we were unable to find genome-wide differences that passed the threshold for genome-wide statistical significance. Among other possible explanations for this fact, one should note the significant inter-individual variability in 5hmC levels observed here and in other brain studies.14, 20, 21, 22, 52 Interestingly, inter-individual variation, although not necessarily to the same extent as 5hmC, also exists for 5mC levels in mammalian brain.15, 53, 54, 55 However, based on previous established criteria for genome-wide significance of genetic variation studies,37 we identified several suggestive 5hmC genomic loci. Of these, we further investigated MYO16 and IDE (suggestive P=1.26 × 10−4 and P=3.9 × 10−4, respectively), both of which show changes in gene expression when groups were compared. Finally, in this study, we used post-mortem brain tissue from caucasian males of French-Canadian descent. Future work should attempt to determine the exact functional role of 5hmC in regulating transcription of genes involved in these disorders, both in the specific population studied here, as well as in other populations showing increased risk of MDD and suicide. This will allow us to better identify avenues of therapeutic intervention, with the hope of better treating depression and preventing suicide.
References
World Health Statistics. World Health Organization. 2016; http://www.who.int.
Ernst C, Mechawar N, Turecki G . Suicide neurobiology. Prog Neurobiol 2009; 89: 315–333.
Cruceanu C, Kutsarova E, Chen ES, Checknita DR, Nagy C, Lopez JP et al. DNA hypomethylation of Synapsin II CpG islands associates with increased gene expression in bipolar disorder and major depression. BMC Psychiatry 2016; 16: 286.
Lopez JP, Lim R, Cruceanu C, Crapper L, Fasano C, Labonte B et al. miR-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat Med 2014; 20: 764–768.
Maheu M, Lopez JP, Crapper L, Davoli MA, Turecki G, Mechawar N . MicroRNA regulation of central glial cell line-derived neurotrophic factor (GDNF) signalling in depression. Transl Psychiatry 2015; 5: e511.
Nagy C, Torres-Platas SG, Mechawar N, Turecki G . Repression of astrocytic connexins in cortical and subcortical brain regions and prefrontal enrichment of H3K9me3 in depression and suicide. Int J Neuropsychopharmacol 2017; 20: 50–57.
Turecki G . The molecular bases of the suicidal brain. Nat Rev Neurosci 2014; 15: 802–816.
Kriaucionis S, Heintz N . The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324: 929–930.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324: 930–935.
Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y . Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010; 466: 1129–1133.
Kinney SM, Chin HG, Vaisvila R, Bitinaite J, Zheng Y, Esteve PO et al. Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J Biol Chem 2011; 286: 24685–24693.
Li W, Liu M . Distribution of 5-hydroxymethylcytosine in different human tissues. J Nucleic Acids 2011; 2011: 870726.
Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res 2012; 22: 467–477.
Gross JA, Pacis A, Chen GG, Barreiro LB, Ernst C, Turecki G . Characterizing 5-hydroxymethylcytosine in human prefrontal cortex at single base resolution. BMC Genomics 2015; 16: 672.
Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341: 1237905.
Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N . MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012; 151: 1417–1430.
Wen L, Li X, Yan L, Tan Y, Li R, Zhao Y et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol 2014; 15: R49.
Coppieters N, Dieriks BV, Lill C, Faull RL, Curtis MA, Dragunow M . Global changes in DNA methylation and hydroxymethylation in Alzheimer's disease human brain. Neurobiol Aging 2014; 35: 1334–1344.
Villar-Menendez I, Blanch M, Tyebji S, Pereira-Veiga T, Albasanz JL, Martin M et al. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington's disease. Neuromolecular Med 2013; 15: 295–309.
Al-Mahdawi S, Virmouni SA, Pook MA . The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front Neurosci 2014; 8: 397.
Ivanov M, Kals M, Lauschke V, Barragan I, Ewels P, Kaller M et al. Single base resolution analysis of 5-hydroxymethylcytosine in 188 human genes: implications for hepatic gene expression. Nucleic Acids Res 2016; 44: 6756–6769.
Lunnon K, Hannon E, Smith RG, Dempster E, Wong C, Burrage J et al. Variation in 5-hydroxymethylcytosine across human cortex and cerebellum. Genome Biol 2016; 17: 27.
Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012; 336: 934–937.
Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012; 149: 1368–1380.
Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 2011; 29: 68–72.
Fiori LM, Gross JA, Turecki G . Effects of histone modifications on increased expression of polyamine biosynthetic genes in suicide. Int J Neuropsychopharmacol 2011; 15: 1161–1166.
Gross JA, Fiori LM, Labonte B, Lopez JP, Turecki G . Effects of promoter methylation on increased expression of polyamine biosynthetic genes in suicide. J Psychiatr Res 2012; 47: 513–519.
Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci USA 2010; 107: 8824–8829.
Cruceanu C, Alda M, Nagy C, Freemantle E, Rouleau GA, Turecki G . H3K4 tri-methylation in synapsin genes leads to different expression patterns in bipolar disorder and major depression. Int J neuropsychopharmacol 2013; 16: 289–299.
Nagy C, Suderman M, Yang J, Szyf M, Mechawar N, Ernst C et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry 2014; 20: 320–328.
Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet 2012; 90: 260–272.
Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry 2012; 69: 314–324.
Dumais A, Lesage AD, Lalovic A, Seguin M, Tousignant M, Chawky N et al. Is violent method of suicide a behavioral marker of lifetime aggression? Am J Psychiatry 2005; 162: 1375–1378.
Sun Z, Terragni J, Borgaro JG, Liu Y, Yu L, Guan S et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep 2013; 3: 567–576.
Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 2013; 152: 642–654.
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2010; 107: 21931–21936.
Lander E, Kruglyak L . Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 1995; 11: 241–247.
Patel KG, Liu C, Cameron PL, Cameron RS . Myr 8, a novel unconventional myosin expressed during brain development associates with the protein phosphatase catalytic subunits 1alpha and 1gamma1. J Neurosci 2001; 21: 7954–7968.
Yokoyama K, Tezuka T, Kotani M, Nakazawa T, Hoshina N, Shimoda Y et al. NYAP: a phosphoprotein family that links PI3K to WAVE1 signalling in neurons. EMBO J 2011; 30: 4739–4754.
Rodriguez-Murillo L, Xu B, Roos JL, Abecasis GR, Gogos JA, Karayiorgou M . Fine mapping on chromosome 13q32-34 and brain expression analysis implicates MYO16 in schizophrenia. Neuropsychopharmacology 2014; 39: 934–943.
Waite K, Eickholt BJ . The neurodevelopmental implications of PI3K signaling. Curr Top Microbiol Immunol 2010; 346: 245–265.
Yfanti C, Mengele K, Gkazepis A, Weirich G, Giersig C, Kuo WL et al. Expression of metalloprotease insulin-degrading enzyme insulysin in normal and malignant human tissues. Int J Mol Med 2008; 22: 421–431.
Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev 2011; 25: 679–684.
Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A, Grayson DR . Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl Psychiatry 2014; 4: e349.
Dong E, Gavin DP, Chen Y, Davis J . Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry 2012; 2: e159.
Kurochkin IV, Goto S . Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett 1994; 345: 33–37.
Yui D, Nishida Y, Nishina T, Mogushi K, Tajiri M, Ishibashi S et al. Enhanced phospholipase A2 group 3 expression by oxidative stress decreases the insulin-degrading enzyme. PLoS ONE 2015; 10: e0143518.
Accardi MV, Brown PM, Miraucourt LS, Orser BA, Bowie D . alpha6-containing GABAA receptors are the principal mediators of inhibitory synapse strengthening by insulin in cerebellar granule cells. J Neurosci 2015; 35: 9676–9688.
Popa-Wagner A, Mitran S, Sivanesan S, Chang E, Buga AM . ROS and brain diseases: the good, the bad, and the ugly. Oxid Med Cell Longev 2013; 2013: 963520.
Tobe EH . Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr Dis Treat 2013; 9: 567–573.
Bakunina N, Pariante CM, Zunszain PA . Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015; 144: 365–373.
Brasa S, Mueller A, Jacquemont S, Hahne F, Rozenberg I, Peters T et al. Reciprocal changes in DNA methylation and hydroxymethylation and a broad repressive epigenetic switch characterize FMR1 transcriptional silencing in fragile X syndrome. Clin Epigenetics 2016; 8: 15.
Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13: R43.
Li X, Baker-Andresen D, Zhao Q, Marshall V, Bredy TW . Methyl CpG binding domain ultra-sequencing: a novel method for identifying inter-individual and cell-type-specific variation in DNA methylation. Genes Brain Behav 2014; 13: 721–731.
Xin Y, Chanrion B, Liu MM, Galfalvy H, Costa R, Ilievski B et al. Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS ONE 2010; 5: e11357.
Acknowledgements
This work was supported by grants to GT from the Canadian Institute of Health Research (MOP93775, MOP11260, MOP119429 and MOP119430), the National Institutes of Health (1R01DA033684) and an investigator-initiated grant from Pfizer Canada, as well as by the Fonds de Recherche du Québec - Santé through a Chercheur National salary award and through the Quebec Network on Suicide, Mood Disorders and Related Disorders. JAG is supported by a CIHR Frederick Banting and Charles Best Doctoral fellowship. PEL is supported by the Fondation Fyssen, the Canadian Institutes of Health Research, the American Foundation for Suicide Prevention, the Fondation Deniker and the Fondation pour la Recherche Médicale.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Gross, J., Pacis, A., Chen, G. et al. Gene-body 5-hydroxymethylation is associated with gene expression changes in the prefrontal cortex of depressed individuals. Transl Psychiatry 7, e1119 (2017). https://doi.org/10.1038/tp.2017.93
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.93
This article is cited by
-
Genome-wide 5-hydroxymethylcytosines in circulating cell-free DNA as noninvasive diagnostic markers for gastric cancer
Gastric Cancer (2024)
-
Genome-wide hydroxymethylation profiles in liver of female Nile tilapia with distinct growth performance
Scientific Data (2023)
-
Post-stroke depression: epigenetic and epitranscriptomic modifications and their interplay with gut microbiota
Molecular Psychiatry (2023)
-
The basolateral amygdala-anterior cingulate pathway contributes to depression-like behaviors and comorbidity with chronic pain behaviors in male mice
Nature Communications (2023)
-
Profiling neuronal methylome and hydroxymethylome of opioid use disorder in the human orbitofrontal cortex
Nature Communications (2023)
