
Abstract
Although it is unclear which cellular factor(s) is responsible for the genetic instability associated with initiating and sustaining cell transformation, it is known that many cancers have mutations that inactivate the Rb-mediated proliferation pathway. We show here that pRb inactivation and the resultant deregulation of one E2F family member, E2F1, leads to DNA double-strand break (DSB) accumulation in normal diploid human cells. These DSBs occur independent of Atm, p53, caspases, reactive oxygen species, and apoptosis. Moreover, E2F1 does not contribute to c-Myc-associated DSBs, indicating that the DSBs associated with these oncoproteins arise through distinct pathways. We also find E2F1-associated DSBs in an Rb mutated cancer cell line in the absence of an exogenous DSB stimulus. These basal, E2F1-associated DSBs are not observed in a p16ink4a inactivated cancer cell line that retains functional pRb, unless pRb is depleted. Thus, Rb status is key to regulating both the proliferation promoting functions associated with E2F and for preventing DNA damage accumulation if E2F1 becomes deregulated. Taken together, these data suggest that loss of Rb creates strong selective pressure, via DSB accumulation, for inactivating p53 mutations and that E2F1 contributes to the genetic instability associated with transformation and tumorigenesis.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others
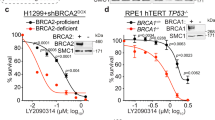


References
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K et al. (2005). Nature 434: 864–870.
Berkovich E, Ginsberg D . (2003). Oncogene 22: 161–167.
Biroccio A, Benassi B, Amodei S, Gabellini C, Del Bufalo D, Zupi G . (2001). Mol Pharmacol 60: 174–182.
Bosco EE . (2004). Nucleic Acids Res 32: 25–34.
DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR . (1997). Proc Natl Acad Sci USA 94: 7245–7250.
Duensing S, Munger K . (2002). Cancer Res 62: 7075–7082.
Dyson N . (1998). Genes Dev 12: 2245–2262.
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T et al. (2005). Nature 434: 907–913.
Johnson DG, Schwarz JK, Cress WD, Nevins JR . (1993). Nature 365: 349–352.
Kowalik TF, DeGregori J, Schwarz JK, Nevins JR . (1995). J Virol 69: 2491–2500.
Leone G, Sears R, Huang E, Rempel R, Nuckolls F, Park CH et al. (2001). Mol Cell 8: 105–113.
Lomazzi M, Moroni MC, Jensen MR, Frittoli E, Helin K . (2002). Nat Genet 31: 190–194.
Matsumura I, Tanaka H, Kanakura Y . (2003). Cell Cycle 2: 333–338.
Morgenbesser SD, Williams BO, Jacks T, DePinho RA . (1994). Nature 371: 72–74.
Nevins JR . (2001). Hum Mol Genet 10: 699–703.
Olive PL, Wlodek D, Banath JP . (1991). Cancer Res 51: 4671–4676.
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM . (2000). Curr Biol 10: 886–895.
Powers JT, Hong S, Mayhew CN, Rogers PM, Knudsen ES, Johnson DG . (2004). Mol Cancer Res 2: 203–214.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM . (1998). J Biol Chem 273: 5858–5868.
Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S et al. (2004). Mol Cell Biol 24: 2968–2977.
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S . (2004). Annu Rev Biochem 73: 39–85.
Schwarz JK, Bassing CH, Kovesdi I, Datto MB, Blazing M, George S et al. (1995). Proc Natl Acad Sci USA 92: 483–487.
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR . (2000). Genes Dev 14: 2501–2514.
Tanaka H, Matsumura I, Ezoe S, Satoh Y, Sakamaki T, Albanese C et al. (2002). Mol Cell 9: 1017–1029.
Tominaga K, Morisaki H, Kaneko Y, Fujimoto A, Tanaka T, Ohtsubo M et al. (1999). J Biol Chem 274: 31463–31467.
Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM et al. (2002). Mol Cell 9: 1031–1044.
White AE, Livanos EM, Tlsty TD . (1994). Genes Dev 8: 666–677.
Acknowledgements
We thank Rachel Gerstein, Kendall Knight, Dario Altieri, Steve Grossman, Nick Rhind, and Kowalik lab members for commenting on the manuscript. This work was supported by National Institutes of Health (NIH) Grant CA86038 (TFK).
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pickering, M., Kowalik, T. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene 25, 746–755 (2006). https://doi.org/10.1038/sj.onc.1209103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1209103
Keywords
This article is cited by
-
DNA damage response(DDR): a link between cellular senescence and human cytomegalovirus
Virology Journal (2023)
-
miR-181a initiates and perpetuates oncogenic transformation through the regulation of innate immune signaling
Nature Communications (2020)
-
Acquired genetic changes in human pluripotent stem cells: origins and consequences
Nature Reviews Molecular Cell Biology (2020)
-
Functional genomics identifies new synergistic therapies for retinoblastoma
Oncogene (2020)
-
Autophagy suppression enhances DNA damage and cell death upon treatment with PARP inhibitor Niraparib in laryngeal squamous cell carcinoma
Applied Microbiology and Biotechnology (2019)
