
Abstract
The mRNAs stored in eggs are crucial for embryogenesis. To address functions of maternal mRNAs, we recently reported the novel method MASK (maternal mRNA-specific knockdown), which we used to specifically knockdown maternal transcripts in the ascidian Ciona intestinalis Type A. In MASK, the cis element of a maternal gene is fused with eGFP or Kaede reporter gene, and the cassette is introduced into Ciona genome by transposon-mediated transgenesis. In eggs of the transgenic lines, the maternal expression of the gene whose cis element is used for driving the reporter gene is suppressed. The zygotic expression of the gene is not suppressed, suggesting that the MASK method can distinguish between maternal and zygotic functions of a gene. Here we investigated the cis and trans factors responsible for MASK results. In the ovaries in which knockdown of a maternal gene occurs, a number of antisense small RNAs are expressed that are complementary to the sequence of the knocked-down genes. We suspect that these antisense small RNAs are the factor responsible for MASK results. The antisense small RNAs have several features that are seen in PIWI-interacting RNAs (piRNAs), suggesting that MASK is likely to use a piRNA-mediated mechanism to knock down maternal mRNAs.
Similar content being viewed by others


Introduction
The initiative cues for animal development are given by genetic substances that are stored in eggs. The characterization of the functions of these maternal factors such as mRNAs and proteins is thus essential for understanding the mechanisms of animal development. The chordate ascidians comprise a well-known animal group, and the involvement of maternal substances on the embryogenesis of chordate ascidians was first described over a century ago1. Molecular studies have characterized the genes whose mRNAs function as the determinative factors for cell differentiation of ascidians2. Maternal factors of ascidians have various functions that are not limited to cell differentiation. These maternal factors are involved in the localization of mRNAs, unequal cleavages of blastomeres, and gastrulation2,3,4,5,6,7,8.
Many studies have revealed that a number of maternal transcripts are localized to the specific region of ascidian eggs9,10,11,12,13,14,15,16,17,18,19, suggesting that these localized RNAs are crucial for embryogenesis. However, the functions of maternally expressed genes have not been characterized well due to technical issues that are not limited to ascidians. First, the knockdown experiments in model organisms that apply antisense technologies are usually not informative for maternal proteins that are already translated during oogenesis. Second, the generation of mutant lines for maternally expressed genes requires an additional generation to observe the appearance of phenotypes compared to the investigation of zygotic functions of genes, because eggs laid by homozygous mutant females are necessary for the examination of the effect of mutations on maternal genes. Third, the generation of homozygous mutant females becomes difficult when the target maternal gene has an essential function for viability as its zygotic function, and thus the knockouts of such maternal genes by genome editing techniques or conventional knockout vectors is frequently not applicable to the phenotypes of maternal functions of the target genes without the use of a special technique such as conditional knockouts. In order to avoid these obstacles, developmental biologists have attempted to improve the methods for analyzing maternally expressed genes [e.g. ref.20,21].
Our group recently established a new reverse genetic method for knocking down the maternal expression of genes in the ascidian Ciona intestinalis Type A22. The method, named MASK (maternal mRNA-specific knockdown), uses an epigenetic suppression of the maternal expression of reporter genes (such as enhanced green fluorescent protein [eGFP] gene) from the cis elements of maternally expressed genes. In Ciona, germ-line transgenesis has been achieved with the Tc1/mariner superfamily transposon Minos23,24,25. In the transgenic lines, the eGFP expression in oocytes and eggs is somehow silenced by an epigenetic mechanism. When the cis element plus the 5′ untranslated region (5′ UTR) of a maternal gene is used to drive eGFP, the maternal expression of the gene is silenced together with eGFP in oocytes and eggs22. Curiously, the zygotic expression of neither eGFP nor the targeted maternal gene (if it has zygotic expression in Ciona) is silenced by MASK. This is a strong advantage for establishing mutant lines of genes that have crucial roles in both maternal and zygotic functions by a reverse genetic method, and for precisely distinguishing between maternal and zygotic functions of genes.
In light of these advantages, the use of MASK is expected to energize the studies of maternal genes in Ciona. A more thorough understanding of the mechanisms that underlie the MASK method will lead to the improvement of the method and to the introduction of MASK to other organisms. However, our understanding of the mechanisms of MASK is limited, and several questions remain to be solved.
In our previous report about the use of MASK22, we induced MASK by eGFP26 and Kaede27 reporter genes, suggesting the possibility that multiple reporter genes could be used for MASK. A deeper investigation of the reporter genes that are compatible with MASK is necessary. In a related matter, the requirement of the vector element necessary to induce MASK must be characterized in order to know how much we can modify the vector for MASK without losing the activity to knockdown maternal gene. It is also unknown how the insertion of the MASK vectors into the Ciona genome leads to the suppression of the maternal expression of endogenous genes. Even though the same MASK vector is used, the degree of knockdowns differs among transgenic lines, and not all of the transgenic lines harboring the MASK vector exhibit a knockdown of the target gene. The latter fact suggests that a certain manner of transgene insertion (perhaps involving the property of the inserted genomic sites or the formation of a concatemer of a transgene) may influence the occurrence of MASK. The uncertain activation of MASK is a serious disadvantage of this method, because screening for an appropriate transgenic line that shows efficient knockdown of the target gene must be conducted.
In this study, we address the characterization of cis and trans factors responsible for the knockdown of the maternal expression of genes by MASK. Our findings demonstrate that there is not a specific element in the vector that is necessary for MASK, suggesting the flexibility of vector design for this method. We also observed that small RNAs that are complementary to the target genes are expressed from the MASK vectors. Several lines of evidence suggest that the antisense small RNAs produced from the MASK vectors are the trans factors responsible for the knockdown of maternal expression in MASK. Based on several characteristics of the small RNAs observed in this study, we deduced the possible pathway creating the small RNAs. This information will be useful for the future improvement of MASK.
Results
Requirement of reporter gene for the maternal-specific knockdown
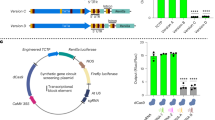
In a previous study, we showed that both eGFP and Kaede can induce MASK22. These reporter genes were driven by the cis elements of targeted maternal genes for inducing MASK. In addition to the MASK cassette, the 1st generation of the MASK vector contained a selectable marker cassette that drives eGFP in the muscle (Fig. 1a)28. Although we showed that the marker cassette could be changed to the one expressing DsRed, the tested vector used eGFP for knocking down the maternal gene (Fig. 1b). Therefore, all of the MASK vectors tested so far harbored eGFP as a genetic element, and this could be a shared feature inducing MASK. In the present study, we extensively analyzed whether eGFP is required for MASK. For this purpose, we modified the design of MASK vectors that target Ci-pem10.

The occurrence of MASK does not rely on the primary structure of reporter gene. (a) The MASK vectors for knocking down Ci-pem. These vectors have the transcriptional cassette expressing eGFP in the muscle (Muscle cassette). ITR, inverted repeat of transposon; NLS, nuclear localization signal sequence; Ter, transcription termination sequence; UTR untranslated region. The colors of the genetic elements correspond to those in Fig. 3. (b) The MASK vector using DsRed cassette as the marker of transgenic lines. Fr3 enhancer, the enhancer isolated from the intron of Ci-Musashi30. (c) The knockdown of Ci-pem. Left panels: The morphology at the larval stage. Normal Ciona larvae exhibit the tadpole morphology. The anteroposterior axis of larvae could not be recognized in the Ci-pem knockdown larvae. Right panels: The expression of maternal Ci-pem mRNA in the unfertilized eggs, as revealed by in situ hybridization. Dark purple color corresponds to Ci-pem mRNA. The abbreviated names of transgenic lines indicate the reporter gene used for knocking down Ci-pem in the transgenic vectors shown in (b). wtGFP, wild-type GFP.
First, the marker cassette that drives DsRed was chosen29 under the control of the promoter of Ci-TPO and the Fr3 enhancer of Ci-Msi30. Second, we exchanged the reporter gene that is driven by Ci-pem cis element with ones other than eGFP. As shown in Fig. 1b, the new vectors do not contain eGFP. We made transgenic lines that have the MASK vectors by using the Minos transposon-mediated system, and we examined whether Ci-pem was or was not knocked down in eggs of the transgenic animals. We observed that Kaede, DsRed, monomeric Kusabira Orange 2 (mKO2)31, and wild-type GFP (the original GFP gene isolated from Aequorea victoria)32 exhibited the knockdown of Ci-pem (Fig. 1, Suppl. Table S1), suggesting that eGFP is not the genetic element required for inducing MASK. MASK can be caused by reporter genes with various DNA sequences.
Requirement of transposon element for MASK
Because the DNA sequence of the reporter gene is not confined to a specific gene for inducing MASK, we next attempted to characterize the vector element other than reporter gene that is necessary for MASK. Because transposable elements are the representative target of epigenetic silencing33, we examined whether Minos transposon element is necessary for MASK. In our previous research, we introduced Sleeping Beauty (SB) transposon-based transgenesis in Ciona34,35,36. We used SB instead of Minos to create transgenic lines for the knockdown of Ci-pem by MASK (Fig. 2a). Two transgenic lines were created, and one exhibited the knockdown of Ci-pem (Fig. 2b), suggesting that Minos is not the factor responsible for MASK.

Transposon element is not required for inducing MASK. (a) The vectors used to analyze the necessity of transposable elements for MASK. SB, sleeping beauty. T2 indicates the improved isoform of SB. (b) Occurrence of Ci-pem knockdown in eggs of the transgenic line created by the sleeping beauty transposon vector. (c) Occurrence of Ci-pem knockdown in eggs of the transgenic line created by the vector (pSPFr3dTPOR;CipemG). Which does not have a transposable element.
To further examine the necessity of transposon element for MASK, we created transgenic lines without transposon elements. We showed previously that the germ-line transformation of Ciona can be achieved by the electroporation of plasmid vectors that do not include a transposon element37. By this electroporation-mediated method, we created two transgenic lines of the vector, pSPFr3dTPOR;CipemG, which did not have a transposon element (Fig. 2a). Among them, one transgenic line exhibited the knockdown of Ci-pem in eggs (Fig. 2c), suggesting that transposon element is not necessary for inducing MASK. Overall, our results showed that transposable element is not required for MASK, confirming that the vector design can be changed flexibly in this method.
Small RNAs complementary to target mRNA are expressed in the ovary of MASK lines
In MASK, the location of the MASK vector in the genome is usually not close to the genomic locus where the target gene is encoded22. A factor created from the MASK vector is thus suspected to reach the target gene or its mRNA to suppress it. To examine whether such a trans factor is present, we sequenced small RNAs isolated from ovaries of the MASK lines, because small RNAs are good candidates for the epigenetic silencing of genes38,39,40. Small RNAs isolated from the ovary of the MASK transgenic line Tg[MiCiTnIGCipemG]2 (this line exhibits silencing of eGFP and Ci-pem in all eggs22) were sequenced, and the small RNAs homologous to the DNA sequence of the Ci-pem MASK vector were mapped onto the vector (Fig. 3a).

Small RNAs that have the homologous sequence to MASK vectors are formed in the ovary of MASK transgenic lines. (a) The small RNAs found in the ovary of Tg[MiCiTnIGCipemG]2. The number at the bottom is the length of the vector in base pairs (bp). The graph shows the number of times (read per million) each nucleotide in the vector appears in the sequenced small RNAs. The red and blue bars in the panels correspond to the results of antisense and sense strands, respectively. The colors correspond to the genetic elements in the vector. The MASK vector contains two identical copies of NLS-eGFP, Ter, and Minos ITRs elements, and we could not distinguish which copy the small RNAs corresponding to the elements are derived from. Therefore, these elements have the same peak patterns between the two copies. (b) The small RNAs found in the ovary of Tg[MiCiNutG]3.
The results revealed the extensive expression of small RNAs that are complementary to the protein coding region (the ORF) of eGFP. In addition, small RNAs that are complementary to the 5′ UTR of Ci-pem, the required element for the knockdown of Ci-pem22, were also extensively expressed (Fig. 3a, Suppl. Table S2). Small RNAs that correspond to the sense strands of eGFP ORF or Ci-pem 5′ UTR were also identified; however, their quantities are much lower than those of antisense small RNAs (blue vs. red lines in Fig. 3a). The peaks of the small RNAs are accumulated around the genetic elements of the vector which is transcribed from the cis elements (Fig. 3), suggesting the necessity of transcription to the production of small RNAs.
The quantities of antisense small RNAs corresponding to the target genes of MASK correlate with the degree of the knockdown of target genes in different MASK lines. A lesser production of eGFP and Ci-pem 5′ UTR antisense small RNAs was observed in the ovary of Tg[MiCiTnIGCipemG]1 compared to that of Tg[MiCiTnIGCipemG]2 (Suppl. Table S2). Tg[MiCiTnIGCipemG]1 exhibited the knockdown of eGFP and Ci-pem in an imperfect manner (some of the eggs of this line escaped from the knockdown of these genes)22. Moreover, when we sequenced small RNAs expressed in the ovary of the transgenic line Tg[MiCiTnIGCipemG]9, which has the same transgene as Tg[MiCiTnIGCipemG]2 but does not exhibit the knockdown of eGFP or Ci-pem22, antisense small RNAs corresponding to eGFP and the Ci-pem 5′ UTR were barely found (Suppl. Table S2).
We sequenced the small RNAs isolated from the somatic tissue (mantle layer) of Tg[MiCiTnIGCipemG]2, where MASK does not occur22. This experiment showed that the expression of the small RNAs corresponding to Ci-pem 5′ UTR was not detected in the somatic tissue of the MASK transgenic line, whereas a considerable amount of small RNAs corresponding to the ORF of eGFP was expressed in the mantle layer (Suppl. Table S2, Tg[MiCiTnIGCipemG]2 mantle). The small RNAs for the eGFP ORF expressed in the mantle of Tg[MiCiTnIGCipemG]2 were very short (they have a peak around 6–10 nt long; Table 1), and they may not have the activity to suppress eGFP, as we discuss later regarding the length of small RNAs effective for the knockdown in MASK.
To further investigate whether the antisense small RNAs are the substance responsible for MASK, we sequenced small RNAs isolated from the transgenic line (Tg[MiCiNutG]3), which exhibits maternal knockdowns of both eGFP and Ci-Nut41 but not Ci-pem22. The ovary of Tg[MiCiNutG]3 expressed abundant antisense small RNAs for eGFP and the Ci-Nut 5′ UTR (Fig. 3b, Suppl. Table S2). This transgenic line also exhibited abundant expression of sense small RNAs for the 5′ UTR of Ci-Nut. Expression of the antisense small RNAs for eGFP and the Ci-Nut 5′ UTR was not seen in the ovary of the other transgenic line Tg[MiCiNutG]4, which has the same transgene as Tg[MiCiNutG]3 but does not show the knockdowns of eGFP or Ci-Nut (Suppl. Table S2)22. In both Tg[MiCiNutG]3 and Tg[MiCiNutG]4, the antisense small RNAs for the Ci-pem 5′ UTR were not seen, suggesting that the creation of antisense small RNAs for a target gene is dependent on the transgene. In conclusion, the production of antisense small RNAs coincides with the occurrence and degree of the knockdown of target genes, suggesting that the small RNAs are the substance responsible for MASK.
The characteristics of MaskRNAs
The region of the eGFP ORF from which eGFP antisense small RNAs are likely to originate is similar between the antisense small RNAs in the ovaries of Tg[MiCiTnIGCipemG]2 and Tg[MiCiTnIGCipemG]1 (Fig. 4a). Moreover, the eGFP antisense small RNAs in the ovaries of Tg[MiCiTnIGCipemG]2 and Tg[MiCiNutG]3 are also formed from similar eGFP regions (Fig. 4b), even though these two transgenic lines drive eGFP from different cis elements. These data suggest that antisense small RNAs for eGFP are formed through a related process in the MASK transgenic lines that is dependent on the primary structure of eGFP.

The comparisons of antisense small RNAs corresponding to the reporter genes. To enhance the visibility, the scales of the vertical bar (corresponding to RPM) are not the same between the graphs. (a) Comparison of eGFP antisense small RNAs between Tg[MiCiTnIGCipemG]2 and Tg[MiCiTnIGCipemG]1. (b) Comparison of eGFP antisense small RNAs between Tg[MiCiTnIGCipemG]2 and Tg[MiCiNutG]3. (c) Comparison of eGFP and Kaede antisense small RNAs between Tg[MiCiTnIGCipemG]2 and Tg[MiFr3dTPORCipemK]4. Because eGFP and Kaede have similar but different nucleotide lengths, the horizontal axis is somewhat different between the two results.
To further examine whether the position of antisense small RNAs is dependent on the sequence of the gene and is independent of the maternal cis element that drives reporter genes, we sequenced the small RNAs isolated from the ovary of Tg[MiFr3dTPORCipemK]4 (Suppl. Table S3). This transgenic line uses Kaede reporter gene for knocking down Ci-pem. Unlike eGFP antisense small RNAs (which are located mostly in the 5′ half of the eGFP ORF), the antisense small RNAs for Kaede were preferentially derived from the region near the 3′ end of Kaede ORF (Fig. 4c). Therefore, the location of the antisense small RNAs is not determined by the cis element adjacent to the reporter gene. Rather, the location of the antisense small RNAs created in the MASK lines is likely to be dependent on the transcribed sequences.
The length of the antisense small RNAs corresponding to the eGFP and Ci-pem 5′ UTR has a peak that is approx. 26–30 nucleotide (nt) long, and their average lengths are 26.0–28.5 nt (Table 1, Suppl. Table S4). The length of the antisense small RNAs is similar to that seen in piRNAs42,43. Moreover, the 5′ end of these antisense RNAs is likely to be uridine (U; Table 2). This characteristic is also seen in piRNAs. The antisense small RNAs are likely to be responsible for MASK, and we thus named them MaskRNAs. The definition of MaskRNAs is: (1) derived from the antisense strand of targeted genes of MASK, (2) the length is approx. 26–30 nt long, and (3) the 1st nucleoside is preferentially U.
We identified the expression of antisense small RNAs that can target the ORF of Ci-pem in the ovary of Tg[MiCiTnIGCipemG]2, Tg[MiCiTnIGCipemG]1 and Tg[MiFr3dTPORCipemK]4 (Suppl. Tables S2, S3). Likewise, antisense small RNAs that can target the ORF of Ci-Nut were also observed in the ovary of Tg[MiCiNutG]3; in the case of Tg[MiCiNutG]3, sense small RNAs that correspond to the ORF of Ci-Nut are more abundant than those for the antisense strand. Because ORFs of Ci-pem or Ci-Nut are not included in the MASK vectors, the presence of the antisense small RNAs of these ORFs suggests that the antisense strands of these ORFs are somehow transcribed in the transgenic lines. Like MaskRNAs, the antisense small RNAs corresponding to the ORFs of targeted genes tend to start with U, and tend to be approx. 26–30 nt long (Tables 1 and 2), suggesting that these antisense small RNAs for ORFs are formed via a process that is similar to that of MaskRNAs.
MASK induces the degradation of target mRNA
There are several processes of target gene downregulations by antisense small RNAs including piRNAs42; i.e., transcriptional silencing, the blocking of translation without degradation, and the degradation of target RNAs. To investigate which mechanism is used in MASK, we observed eGFP mRNA microinjected into eggs derived from MASK-positive and MASK-negative (control) animals. To mimic the occurrence of MASK, eGFP mRNA was microinjected into unfertilized eggs and then the eggs were subjected to fertilization to develop until the tailbud stage. We observed the fluorescence of eGFP expressed in the tailbud embryos, and detected eGFP mRNAs in the embryos by means of in situ hybridization.
The results revealed that the injected eGFP mRNA remained to be translated throughout the body of the control animals developed from wild-type eggs × sperm of the MASK transgenic line (Tg[MiCiTnIGCipemG]2), whereas the mRNA was almost abolished in embryos developed from wild-type sperm × eggs of the MASK transgenic line (Fig. 5). This suggests that eGFP mRNA is degraded in MASK eggs. Because we keep the MASK transgenic line as hemizygous animals, about one-half of the examined embryos possessed TnI > eGFP cassette that expresses eGFP zygotically in the muscle cells. The zygotic eGFP in the muscle was not suppressed in embryos developed from MASK eggs (Fig. 5b,e). In order to examine whether muscle cells have the ability to suppress maternal eGFP mRNA, we observed the eGFP expression of TnI > eGFP negative embryos into which eGFP mRNA was microinjected before fertilization. The embryos exhibited a reduction of injected eGFP mRNA in all blastomeres including muscle (Fig. 5c,f), suggesting that the muscle cell lineage possesses the ability to downregulate eGFP supplied before fertilization. Therefore, eGFP mRNA expressed in a zygotic manner can somehow escape from MASK.

MASK induces the degradation of mRNA. All displayed embryos are derived from eggs in which eGFP mRNA was microinjected before fertilization. (a–c) Pseudocolored fluorescent images of eGFP. (d–f) eGFP mRNA detected by means of in situ hybridization. Dark purple color illustrates the presence of eGFP mRNA. (a–c) Late tailbud embryos. (d–f) Early tailbud embryos. (a,d) Embryos developed from the cross of wild-type eggs × sperm of Tg[MiCiTnIGCipemG]2. (b,c,e,f) Embryos developed from the cross of wild-type sperm × eggs of Tg[MiCiTnIGCipemG]2. Mu, eGFP signal derived from the zygotic expression in the muscle lineage.
Discussion
In the present study, we addressed how MASK, the maternal specific knockdown, occurs in Ciona to obtain clues for the future improvement of this technique. Our findings demonstrated that there is not a specific DNA stretch in the MASK vectors that is necessary to induce knockdown. Small RNAs that have sequences that are complementary to the target genes are abundantly produced in the ovaries of MASK transgenic lines. Considering that the production of small RNAs is concentrated on the DNA stretches transcribed from the maternal cis element of MASK vectors, the requiring characteristic of MASK vectors is the cis element that can drive reporter gene expression in the maternal fashion. It is likely that the sense and antisense RNAs transcribed from the maternal cis element are used as the seeds to create MaskRNAs (see the discussion below). This transcription-dependent hypothesis can explain the lack of the need for a specific DNA sequence in the MASK vector for inducing MASK.
We observed that antisense small RNAs that are suspected to downregulate target gene are produced in the ovaries of MASK transgenic lines. Among them, we defined MaskRNAs as the small RNAs that have characteristics similar to those of PIWI-interacting (pi)RNAs42,43,44. Indeed, the length of the major antisense small RNAs produced in the ovaries of MASK lines is approx. 26–30 nt long. Typical microRNAs that are known to act to regulate gene expression have uniform lengths, i.e., approx. 21–22 nt45, suggesting that the mechanism underlying the production of MaskRNAs is likely to be different from the mechanism for producing microRNAs. The length of 26–30 nt coincides with the characteristic of piRNAs. The 5′ terminals of antisense piRNAs are preferentially U. The major antisense small RNAs produced in the ovaries of MASK lines also preferentially have U at their 5′ end.
If we assume that MaskRNAs are a type of piRNAs, several characteristics of MASK can be explained. piRNAs are the major RNAs that function in the post-transcriptional silencing of transposons in the gonad, and based on this characteristic, piRNAs are usually expressed specifically in the gonad in mouse46,47,48. The specificity of piRNAs in the gonad coincides with the finding that MASK occurs specifically in oocytes and eggs; the zygotic expression of target genes is not suppressed by MASK. piRNAs are amplified by a ‘ping-pong’ mechanism that requires both sense and antisense strands of the transcripts of target genes (and creates both sense and antisense small RNAs)49. As we stated above in the Results section, the formation of MaskRNAs is concentrated on the maternal transcriptional unit in the examined MASK vectors. This can be explained if we assume that MaskRNAs are produced via the transcription-dependent ping-pong mechanism. We suspect that the requirement of a part of the target gene (in the case of our previous study, the 5′ UTR22) in the transcription unit exists because the genetic element serves as the seed to create piRNAs that can target the target maternal mRNAs. piRNAs are known to cause the degradation of target transcripts43. Our present findings demonstrated that mRNA degradation is a major mechanism of the downregulation of maternal transcripts in MASK.
piRNAs are known to be produced from piRNA clusters44. There are three types of piRNA clusters, classified by the strands of DNAs subjected to transcription. Among them, the uni-strand cluster may not work in Ciona MaskRNA production, because not all transgenic lines of MASK vector exhibited the occurrence of MASK even though they have the same uni-directional transcription unit that transcribes sense strand mRNAs of the target gene and reporter gene (Fig. 6a). Rather, the dual-strand and/or bi-directional cluster may be more appropriate models to explain MASK.

Mechanisms of the occurrence of MASK (maternal mRNA-specific knockdown). (a) Schematic illustration of the occurrence of MASK in the transgenic lines of a MASK vector. (b,c) Concatemers of transgenes in the MASK transgenic lines, as revealed by Southern blotting. Panel b shows the locations of the EcoRV restriction site in the pMiCiTnIGCipemG vector (see Fig. 1a) that was used to create Tg[MiCiTnIGCipemG]1, as an example of the experiment in panel c. Because the eGFP probe can hybridize with the left and right fragments of this vector (the probe in b), the single insertion of this vector will yield two bands by Southern blotting. The appearance of multiple bands as shown in (c) indicates the insertion of multiple vector elements at the single genomic position, suggesting the formation of concatemers. The examined transgenic lines are shown at the bottom of the panel. Lanes 1 and 2 were detected with eGFP probe, while lanes 3 and 4 were detected with DsRed probe. Tg[Fr3dTPORCipemG]4 and Tg[T2Fr3dTPORCipemG]3 respectively correspond to w/o transposon and Sleeping beauty in Fig. 2.
In Ciona, the insertion of transposons often forms a concatemer of transgenes25,50,51. Such a concatemer may have a chance to mimic a dual-strand and/or bi-directional piRNA cluster by the rearranged transcriptional units (Fig. 6a). Indeed, all MASK-positive transgenic lines (n = 8) possessed a concatemerized transgene, as revealed by Southern blotting (Fig. 6b,c). Our MASK transgenic lines usually possess the transgene insertion at a single genomic site per line. If a single transposon vector were inserted into the insertion site, the transposon insertion would have yielded a limited number of bands by Southern blotting (Fig. 6b). However, the numbers of bands detected by Southern blotting were much greater than the expected numbers, indicating that multiple vector elements were inserted into the single genomic loci, and suggesting the formation of the concatemers. The formation of a piRNA cluster-like genetic element in the Ciona genome probably does not require a specific DNA sequence. This is in accordance with our present results showing that a specific DNA element is unnecessary for MASK vector. Moreover, the creation of an appropriate concatemer for the production of MaskRNAs may occur by chance, suggesting that MASK occurs in some transgenic lines that have MASK vectors as the transgene and that the vector copies are appropriately rearranged in their genome so as to mimic a piRNA cluster. This characteristic can explain why MASK could not be induced in all transgenic lines. The knockdown of genes in other loci by tandem arrays of transgenes was reported in the event named paramutation52. In the animal Drosophila melanogaster, piRNAs are produced from the tandem transgenes, which is essential for the paramutation53. Probably MASK in Ciona uses the mechanism similar to Drosophila paramutation.
The similarity between MaskRNAs and piRNAs suggests that the design of a MASK vector could be improved by mimicking the mechanism producing piRNAs. For example, the transcriptional unit of both sense and antisense strands of a part of the target maternal genes, like that shown in the bottom of Fig. 6, would greatly enhance the occurrence of MASK in all transgenic lines of the MASK vector. We could apply MASK to other tissues of Ciona where piRNAs could be produced. The testis is a strong candidate for such improvement because piRNAs are primarily produced in the testes in mice. In addition, MASK could be introduced into other organisms for facilitating functional analyses of maternal factors, since piRNA is observed in various metazoans54. We would also like to emphasize that the characterization of the mechanisms of a poorly-understood technique is fruitful for advancing the technique, as illustrated by our present findings regarding MASK.
Methods
Constructs
The open reading frames (ORFs) of mKO231 and wild-type GFP32 were polymerase chain reaction (PCR)-amplified. The ORFs were subcloned into the BamHI and EcoRI sites of pSPeGFP25 to create pSPmKO2 and pSPwtGFP. The 5′ upstream region including the 5′ UTR of Ci-pem was isolated by PCR. The PCR fragment was digested with BamHI and subcloned into the BamHI site of pSPKaede55, pSPNLS-DsRed56, pSPmKO2, and pSPwtGFP.
The fusion cassettes were subcloned into pMiFr3dTPORDestR22 using the Gateway® technology (Invitrogen, Carlsbad, CA). Fr3dTPOR cassette was PCR-amplified, and the PCR product was subcloned into the BglII/EcoRV sites of pT2HB57 to create pT2Fr3dTPOR. The Gateway cassette was inserted into the EcoRV site of pT2Fr3dTPOR to create pT2RfB(R)Fr3dTPOR. Ci-pem > NLS::eGFP cassette was subcloned into pT2RfB(R)Fr3dTPOR using the Gateway technology. A Gateway cassette was subcloned into the blunted BglII site of pSPFr3dTPOR56 to create pSPFr3dTPORRfC1. Ci-pem > NLSeGFP cassette was subcloned into pSPFr3dTPORRfC1 using the Gateway technology. The official names of the vectors and transgenic lines according to the nomenclature rules for tunicates58 (Stolfi et al., 2015) were listed in Suppl. Table S5.
Transgenic lines
The transgenic lines were created by transposon-mediated transgenesis or electroporation-mediated transgenesis as described25,36,37. Genomic DNA was isolated from the sperm of transgenic lines. The genomic DNA was digested with EcoRV, and Southern blotting was carried out according to a previous study59.
Microinjection and in situ hybridization
eGFP mRNA was synthesized using the MEGAscript® T3 kit (Ambion, Carlsbad, CA), the Poly(A) Tailing Kit (Ambion), and Cap structure analog (New England Biolabs, Ipswich, MA) as described60. We microinjected eGFP mRNA into unfertilized eggs derived from Tg[MiCiTnIGCipemG]2 or wild-type animals as described61. The microinjected unfertilized eggs were fertilized by sperm of counterpart animals so as to unify the genetic background. The concentration of mRNA in the injection medium was adjusted to 500 ngμl−1. After the embryos were fixed at the appropriate stage, whole-mount in situ hybridization (WISH) was performed as described60,62. The eGFP fluorescence was observed with a fluorescent microscope at the late tailbud stage.
RNA-seq
We surgically isolated ovaries and mantle layers from well-grown Ciona adults of the transgenic lines. The ovaries were mashed with homogenizers in ISOGEN reagent (NipponGene, Tokyo). RNA was extracted according to the manufacturer’s instructions. After treatment with DNaseI, RNAs were subjected to phenol-chloroform and chloroform extraction, and then ethanol-precipitated. A 1-μg aliquot of each precipitated total RNA was then resuspended in 5 μl of nuclease-free water and used to construct a sequence library with the use of a TruSeq Small RNA Sample Prep Kit (Illumina, San Diego, CA) following the manufacturer’s instruction.
The amounts and sizes of the sequence libraries were measured on an Agilent 2100 Bioanalyzer with a DNA 1000 Chip (Agilent Technologies, Santa Clara, CA). Sequencing was performed on a HiSeq. 1500 (Illumina) or Miseq (Illumina) high throughput sequencer using a single-end 100-cycle run. Total reads were extracted with CASAVA v1.8.2 software (Illumina). The obtained sequences were uploaded in the Sequence Read Archive (SRA) (SRA ID: SRR6012511-SRR6012517; Suppl. Table S6).
Next, adaptor sequences and low-quality reads were removed from the extracted reads using the Trimmomatic 0.33 command line tool63. The remaining reads were then aligned using the Bowtie ver. 2.2.3 program64 allowing up to 20 multiple-hits to the reference sequences. The reference sequences were constructed by combining all of the plasmid sequences used for recombination and the C. intestinalis genome (KH, ver. 2008), which was downloaded from the Ghost Database65. The reads per megareads (RPM) value of each nucleotide in reference sequences was calculated by dividing the number of mapped reads (depth) for each nucleotide in the reference sequence calculated using samtools (ver. 1.3)66 by the total reads (M reads). The frequency of first residues and the length of the reads were calculated for the ORF region and the 5′ UTR region of eGFP, Kaede, Ci-pem, and Ci-Nut. The depth and frequency analyses were performed separately for sense RNAs and antisense RNAs.
References
Conklin, E. G. Organ forming substances in the eggs of ascidians. Biol. Bull. 8, 205–230 (1905).
Nishida, H. Specification of embryonic axis and mosaic development in ascidians. Dev. Dyn. 233, 1177–1193 (2005).
Nishida, H. & Sawada, K. macho-1 encodes a localized mRNA in ascidian eggs that specifies muscle fate during embryogenesis. Nature 409, 679–680 (2001).
Nishida, H. Vegetal egg cytoplasm promotes gastrulation and is responsible for specification of vegetal blastomeres in embryos of the ascidian Halocynthia roretzi. Development 122, 1271–1279 (1996).
Nishikata, T., Hibino, T. & Nishida, H. The centrosome-attracting body, microtubule system, and posterior egg cytoplasm are involved in positioning of cleavage planes in the ascidian embryo. Dev Biol 209, 72–85 (1999).
Nakamura, Y., Makabe, K. W. & Nishida, H. POPK-1/Sad-1 kinase is required for the proper translocation of maternal mRNAs and putative germ plasm at the posterior pole of the ascidian embryo. Development 132, 4731–4742 (2005).
Nakamura, Y., Makabe, K. W. & Nishida, H. The functional analysis of Type I postplasmic/PEM mRNAs in embryos of the ascidian Halocynthia roretzi. Dev Genes Evol 216, 69–80 (2006).
Negishi, T., Takada, T., Kawai, N. & Nishida, H. Localized PEM mRNA and protein are involved in cleavage-plane orientation and unequal cell divisions in ascidians. Curr Biol 17, 1014–1025 (2007).
Yoshida, S., Marikawa, Y. & Satoh, N. Posterior end mark, a novel maternal gene encoding a localized factor in the ascidian embryo. Development 122, 2005–2012 (1996).
Yoshida, S., Satou, Y. & Satoh, N. Maternal genes with localized mRNA and pattern formation of the ascidian embryo. Cold Spring Harb. Symp. Quant. Biol. 62, 89–96 (1997).
Satou, Y. & Satoh, N. Posterior end mark 2 (pem-2), pem-4, pem-5, and pem-6: maternal genes with localized mRNA in the ascidian embryo. Dev Biol 192, 467–481 (1997).
Sasakura, Y., Ogasawara, M. & Makabe, K. W. HrWnt-5: a maternally expressed ascidian Wnt gene with posterior localization in early embryos. Int J Dev Biol 42, 573–579 (1998).
Sasakura, Y., Ogasawara, M. & Makabe, K. W. Maternally localized RNA encoding a serine/threonine protein kinase in the ascidian. Halocynthia roretzi. Mech Dev 76, 161–163 (1998).
Satou, Y. posterior end mark 3 (pem-3), an ascidian maternally expressed gene with localized mRNA encodes a protein with Caenorhabditis elegans MEX-3-like KH domains. Dev Biol 212, 337–350 (1999).
Sasakura, Y., Ogasawara, M. & Makabe, K. W. Two pathways of maternal RNA localization at the posterior-vegetal cytoplasm in early ascidian embryos. Dev Biol 220, 365–378 (2000).
Nakamura, Y., Makabe, K. W. & Nishida, H. Localization and expression pattern of type I postplasmic mRNAs in embryos of the ascidian Halocynthia roretzi. Gene Expr Patterns 3, 71–75 (2003).
Yamada, L., Kobayashi, K., Satou, Y. & Satoh, N. Microarray analysis of localization of maternal transcripts in eggs and early embryos of the ascidian. Ciona intestinalis. Dev. Biol. 284, 536–550 (2005).
Yamada, L. Embryonic expression profiles and conserved localization mechanisms of pem/postplasmic mRNAs of two species of ascidian, Ciona intestinalis and Ciona savignyi. Dev Biol 296, 524–536 (2006).
Prodon, F., Yamada, L., Shirae-Kurabayashi, M., Nakamura, Y. & Sasakura, Y. Postplasmic/PEM RNAs: a class of localized maternal mRNAs with multiple roles in cell polarity and development in ascidian embryos. Developmental Dynamics 236, 1698–1715 (2007).
Yoshida, K. et al. Germ cell regeneration-mediated, enhanced mutagenesis in the ascidian Ciona intestinalis reveals flexible germ cell formation from different somatic cells. Dev Biol 423, 111–125 (2017).
Blitz, I. L., Fish, M. B. & Cho, K. W. Leapfrogging: primordial germ cell transplanation permits recovery of CRISPR/Cas9-induced mutations in essential genes. Development 143, 2868–2875 (2016).
Iitsuka, T. et al. Transposon-mediated targeted and specific knockdown of maternally expressed transcripts in the ascidian Ciona intestinalis. Sci Rep 4, 5050 (2014).
Franz, G. & Savakis, C. Minos, a new transposable element from Drosophila hydei, is a member of the Tc1-like family of transposons. Nucleic Acids Res 19, 6646 (1991).
Sasakura, Y., Awazu, S., Chiba, S., Kano, S. & Satoh, N. Application of Minos, one of the Tc1/mariner superfamily transposable elements, to ascidian embryos as a tool for insertional mutagenesis. Gene 308, 11–20 (2003).
Sasakura, Y., Awazu, S., Chiba, S. & Satoh, N. Germ-line transgenesis of the Tc1/mariner superfamily transposon Minos in Ciona intestinalis. Proc. Natl. Acad. Sci. USA 100, 7726–7730 (2003).
Zhang, G., Gurtu, V. & Kain, S. An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res. Commun 227, 707–711 (1996).
Ando, R., Hama, H., Yamamoto-Hino, M., Mizuno, H. & Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci USA 99, 12651–12656 (2002).
Davidson, B. & Levine, M. Evolutionary origins of the vertebrate heart: Specification of the cardiac lineage in Ciona intestinalis. Proc. Natl. Acad. Sci. USA 100, 11469–11473 (2003).
Matz, M. et al. Fluorescent proteins from nonbiolominescent Anthozoa species. Nat Biotechnol 17, 969–973 (1999).
Awazu, S. et al. An enhancer trap in the ascidian Ciona intestinalis identifies enhancers of its Musashi orthologous gene. Dev. Biol. 275, 459–472 (2004).
Kikuchi, A. et al. Structural characterization of a thiazoline-containing chromophore in an orange fluorescent protein, monomeric Kusabira Orange. Biochemistry 47, 11573–11580 (2008).
Prasher, D., Eckenrode, V., Ward, W., Prendergast, F. & Cornier, M. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111, 229-233 (1992).
Slotkin, R. & Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285 (2007).
Ivics, Z., Hackett, P. B., Plasterk, R. H. & Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91, 501–510 (1997).
Mates, L. et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41, 753–761 (2009).
Hozumi, A. et al. Germline transgenesis of the chordate Ciona intestinalis with hyperactive variants of sleeping beauty transposable element. Dev Dyn 242, 30–43 (2013).
Matsuoka, T., Awazu, S., Shoguchi, E., Satoh, N. & Sasakura, Y. Germline transgenesis of the ascidian Ciona intestinalis by electroporation. genesis 41, 61–72 (2005).
Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998).
Bourc’his, D. & Voinnet, O. A small-RNA perspective on gametogenesis, fertilization, and early zygotic development. Science 330, 617–622 (2010).
Meister, G. Argonaute proteins: functional insights and emerging roles. Nat. Rev. Genet. 14, 447–459 (2013).
Etani, K. & Nishikata, T. Novel G-protein-coupled receptor gene expressed specifically in the entire neural tube of the ascidian Ciona intestinalis. Dev Genes Evol 212, 447–451 (2002).
Khurana, J. & Theurkauf, W. piRNAs, transposon silencing, and Drosophila germline development. J. Cell Biol. 191, 905–913 (2010).
Siomi, M. C., Sato, K., Pezic, D. & Aravin, A. A. PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 12, 246–258 (2011).
Yamanaka, S., MC., S. & Siomi, H. piRNA clusters and open chromatin structure. Mobile DNA 5, 22 (2014).
Wheeler, B. et al. The deep evolution of metazoan microRNAs. Evol. Dev. 11, 50–68 (2007).
Aravin, A. et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207 (2006).
Girard, A., Sachidanandam, R., Hannon, G. J. & Carmell, M. A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199–202 (2006).
Grivna, S. T., Beyret, E., Wang, Z. & Lin, H. A novel class of small RNAs in the mouse spermatogenic cells. Genes Dev. 20, 1709–1714 (2006).
Klattenhoff, C. et al. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell 138, 1137–1149 (2006).
Sasakura, Y. et al. Transposon-mediated insertional mutagenesis revealed the functions of animal cellulose synthase in the ascidian Ciona intestinalis. Proc. Natl. Acad. Sci. USA 102, 15134–15139 (2005).
Sasakura, Y. Germline transgenesis and insertional mutagenesis in the ascidian Ciona intestinalis. Dev. Dyn. 236, 1758–1767 (2007).
Hollick, J. B. Paramutation and related phenomena in diverse species. Nat. Rev. Genet. 18, 5–23 (2017).
de Vanssay, A. et al. Paramutation in Drosophila linked to emergence of a piRNA-producing locus. Nature 490, 112–115 (2012).
Palakodeti, D., Smielewska, M., Lu, Y., Yeo, G. & Graveley, B. The PIWI proteins SMEDWI-2 and SMEDWI-3 are required for stem cell function and piRNA expression in planarians. RNA 14, 1174–1186 (2008).
Horie, T. et al. Ependymal cells of chordate larvae are stem-like cells that form the adult nervous system. Nature 469, 525–528 (2011).
Sasakura, Y., Konno, A., Mizuno, K., Satoh, N. & Inaba, K. Enhancer detection in the ascidian Ciona intestinalis with transposase-expressing lines of Minos. Dev. Dyn. 237, 39–50 (2008).
Zayed, H., Izsvak, Z., Khare, D., Heinemann, U. & Ivics, Z. The DNA-bending protein HMGB1 is a cellular cofactor of Sleeping Beauty transposition. Nucleic Acids Res. 31, 2313–2322 (2003).
Stolfi, A. et al. Guideline for the nomenclature of genetic elements in tunicate genomes. Genesis 53, 1–14 (2015).
Hozumi, A. et al. Efficient transposition of a single Minos transposon copy in the genome of the ascidian Ciona intestinalis with a transgenic line expressing transposase in eggs. Dev. Dyn. 239, 1076–1088 (2010).
Sasakura, Y., Suzuki, M. M., Hozumi, A., Inaba, K. & Satoh, N. Maternal factor-mediated epigenetic gene silencing in the ascidian Ciona intestinalis. Mol Genet Genomics 283, 99–110 (2010).
Hikosaka, A., Kusakabe, T., Satoh, N. & Makabe, K. W. Introduction and expression of recombinant genes in ascidian embryos. Dev. Growth Differ. 34, 627–634 (1992).
Yasuo, H. & Satoh, N. An ascidian homolog of the mouse Brachyury (T) gene is expressed exclusively in notochord cells at the fate restricted stage. Dev. Growth Differ. 36, 9–18 (1994).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Satou, Y. et al. Improved genome assembly and evidence-based global gene model set for the chordate Ciona intestinalis: new insight into intron and operon populations. Genome Biol 9, R152 (2008).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Acknowledgements
We thank members of the Shimoda Marine Research Center at the University of Tsukuba for their kind cooperation with our study, and Drs. Shigeki Fujiwara, Yutaka Satou, Manabu Yoshida, and all members of the Maizuru Fishery Research Station of Kyoto University, Misaki Marine Biological Station at the University of Tokyo for the collection of Ciona adults. We are grateful to Prof. Atsushi Miyawaki for the kind provision of wild-type GFP cDNA. This study was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology (MEXT) to Y.S. and T.I. This study was further supported by grants from the National Bioresource Project.
Author information
Authors and Affiliations
Contributions
T.S., T.I. and Y.S. designed the research. T.S., T.I., A.H., A.S. and Y.S. performed the experiments. T.S., T.I., A.H., A.S., H.S. and Y.S. analyzed the data. Y.S. wrote the paper, and T.S., T.I., A.H., A.S. and H.S. contributed in the form of discussions and critical comments.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Satoh, T., Iitsuka, T., Shiraishi, A. et al. piRNA-like small RNAs are responsible for the maternal-specific knockdown in the ascidian Ciona intestinalis Type A. Sci Rep 8, 5869 (2018). https://doi.org/10.1038/s41598-018-24319-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24319-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
