
Abstract
Protective immunity against Mycobacterium tuberculosis depends on the generation of a TH1-type cellular immune response, characterized by the secretion of interferon-γ (IFN-γ) from antigen-specific T cells. The induction of potent cellular immune responses by vaccination in humans has proven difficult. Recombinant viral vectors, especially poxviruses and adenoviruses, are particularly effective at boosting previously primed CD4+ and CD8+ T-cell responses against a number of intracellular pathogens in animal studies1,2,3,4,5,6,7. In the first phase 1 study of any candidate subunit vaccine against tuberculosis, recombinant modified vaccinia virus Ankara (MVA) expressing antigen 85A (MVA85A) was found to induce high levels of antigen-specific IFN-γ-secreting T cells when used alone in bacille Calmette-Guérin (BCG)-naive healthy volunteers. In volunteers who had been vaccinated 0.5–38 years previously with BCG, substantially higher levels of antigen-specific IFN-γ-secreting T cells were induced, and at 24 weeks after vaccination these levels were 5–30 times greater than in vaccinees administered a single BCG vaccination. Boosting vaccinations with MVA85A could offer a practical and efficient strategy for enhancing and prolonging antimycobacterial immunity in tuberculosis-endemic areas.
Similar content being viewed by others


Main
Mycobacterium bovis BCG does not protect against adult pulmonary tuberculosis, but does protect against disseminated disease in childhood when administered at birth in developing countries8,9. Although repeated vaccination with BCG appears not to enhance protection against tuberculosis further10, the protective effect of BCG in childhood might be retained by a vaccination strategy that improved BCG rather than replaced it. The secretion of IFN-γ is central to the activation of M. tuberculosis-infected macrophages, and the measurement of IFN-γ release from antigen-specific T cells provides the best available immunological correlate of protection against tuberculosis11,12,13. HLA class II–restricted CD4+ T cells are essential for protective immunity against M. tuberculosis and class I–restricted CD8+ T cells also have a role14,15,16.
Heterologous prime-boost immunization strategies induce higher levels of effector T-cell responses in animals and humans than homologous boosting with the same vaccine1,2,17. In tuberculosis, incorporating BCG into such a heterologous prime-boost regime retains the protective effects of BCG. Antigen 85A is highly conserved amongst all mycobacterial species and is present in all strains of BCG18. It is immunodominant in animal and human studies, and is protective in small animals5,19,20. The immunogenicity and protective efficacy of boosting BCG with viral vectors expressing antigen 85A in several animal models has previously been documented (ref. 21 and unpublished data).
Here we report the results of a series of phase 1 studies in human volunteers evaluating the safety and immunogenicity of MVA85A, first in BCG-naive healthy volunteers and subsequently in volunteers who have been vaccinated with BCG 0.5–38 years previously.
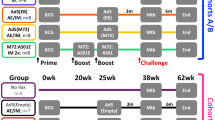
Immunization with MVA85A was safe and well-tolerated (Table 1). The kinetics and magnitude of the antigen-specific T-cell response induced by vaccination with BCG alone, MVA85A alone and BCG prime-MVA85A boost were compared. All three vaccination regimes induced substantial immune responses using purified protein derivative (PPD) from M.tuberculosis, antigen 85 protein or overlapping peptides from antigen 85A as antigen in the assays (Table 2 and Fig. 1). There was a significant main effect of vaccine in the PPD (F = 3.624; P = 0.037), antigen 85 (F = 16.605; P < 0.001) and summed pooled antigen 85A peptide groups (F = 39.982; P < 0.001). Immunization with BCG induced moderate levels of antigen-specific IFN-γ-secreting T cells, which peaked 4 weeks after immunization (Table 2 and Fig. 1b–d). Responses to the pooled antigen 85A peptides were notably weak following BCG vaccination (Fig. 1d). Only 4 of the 11 volunteers responded to any of the 7 peptide pools in the ex vivo ELISPOT assay. These peptide pool responses were all attributable to peptides 12, 13, 27 and 28, and were completely abrogated by CD4+ T-cell depletion.

(a) Timeline for vaccinations (weeks) in each group. (b) Tuberculin PPD responses. (c) Purified antigen 85 protein responses. (d) Summed pooled peptide responses. (e) For each of the three antigens measured, the responses between each vaccine group at each timepoint were compared using the Mann-Whitney statistical test. Statistically significant comparisons are indicated. (f) T-cell epitope display after MVA boost in BCG vaccinated individuals. CD4+ T-cell depletion completely abrogated all individual peptide responses.
In BCG-naive volunteers, a single immunization with MVA85A induced high levels of antigen-specific IFN-γ-secreting T cells, which peaked 7 d after vaccination in 13 of 14 volunteers (Table 2 and Fig. 1b,c). By 4 weeks this response had fallen to a level just above baseline. We did not see a boosting effect of the second vaccination with MVA85A, administered at week 3. One volunteer did not develop insert-specific T cells following immunization with MVA85A; however, this volunteer did develop specific T-cell responses to the MVA vector, despite having no previous history of vaccinia immunization (data not shown).
In contrast to BCG vaccination, vaccination with MVA85A induced strong responses to several peptide pools in 13 of 14 responding volunteers (Table 2 and Fig. 1d). We saw responses to a broad range of peptides across the whole length of antigen 85A. These individual peptide responses were all completely abrogated by CD4+ T-cell depletion (data not shown).
In 16 of 17 volunteers in the BCG prime-MVA85A boost group, a substantial and significant (P < 0.001) rise in antigen-specific T cells was seen 1 week after vaccination (Table 2 and Fig. 1). The peak response 1 week after immunization was significantly higher (P < 0.05) in the BCG prime-MVA85A boost group than in either the BCG or MVA85A alone groups (Fig. 1b–e). These responses were sustained at a significantly higher level than after vaccination with either BCG or MVA85A alone, for at least 24 weeks (Fig. 1e). The baseline responses at screening were higher in the volunteers previously vaccinated with BCG than in the BCG-naive group, as would be expected. Nonetheless, the responses 24 weeks after vaccination with MVA85A in the BCG prime-MVA85A boost group were significantly higher than baseline counts in this group for PPD (Wilcoxon z = −3.010, P = 0.003), antigen 85 (Wilcoxon z = −3.516, P < 0.001) and the summed pooled peptides (Wilcoxon z = −3.408, P = 0.001).
The breadth of peptide responses seen in the MVA85A alone (not shown) and BCG prime-MVA85A boost groups (Fig. 1f) were very similar. But the magnitude of responses was significantly higher (P < 0.05) in the BCG prime-MVA85A boost group (Fig. 1e). Peripheral blood mononuclear cells (PBMC) from 12 volunteers in the BCG prime-MVA85A boost group were assayed with all 66 peptides from antigen 85A. Several of these peptides were recognized by more than 50% of subjects (Fig. 1f), showing the promiscuous recognition of these peptides by different HLA class II molecules, as previously reported19.
The magnitude of T-cell responses seen in the MVA85A alone group are stronger by a factor of about 10 than those seen with other recombinant MVAs used to date17,22. A recombinant MVA expressing an antigen from P. falciparum induced a mean summed peptide response of 90 spot-forming cells (SFC)/1 × 106 PBMC 7 d after vaccination17. In contrast, we saw a mean response to the summed peptides of 1,365 SFC/1 × 106 PBMC 7 d after vaccination with MVA85A (Table 2). One explanation for this is that these volunteers have some pre-existing antimycobacterial immunity that is being boosted by immunization with MVA85A. We used a cultured, rather than an ex vivo ELISPOT assay to investigate this further. The cultured ELISPOT assay has previously been shown to measure central memory T cells, rather than the activated effector T cells that are measured by ex vivo ELISPOT23,24. We performed a cultured IFN-γ ELISPOT assay on the prevaccination screening PBMC from four of the volunteers in the MVA85A alone group. Cells were cultured with M. tuberculosis PPD, M. avium PPD and recombinant antigen 85A. All four volunteers responded to M. avium PPD, two of four responded to M. tuberculosis PPD and two of four responded to recombinant antigen 85A (Fig. 2). None of these volunteers had any baseline responses to either M. tuberculosis PPD or purified antigen 85 on the screening ex vivo ELISPOT.

Screening blood cultured ELISPOT responses to M. tuberculosis PPD; M. avium PPD and recombinant antigen 85A; from four volunteers in the MVA85A-alone study, before vaccination.
These are the first results of the clinical evaluation of any subunit candidate tuberculosis vaccine since BCG was first used over 80 years ago. We observed induction of high levels of antigen-specific T cells when BCG-naive volunteers are immunized with a single low dose of MVA85A. The cultured ELISPOT data presented here show that four of four volunteers tested have pre-existing central memory responses to M. avium PPD, supporting the hypothesis that vaccination with MVA85A is boosting these pre-existing antimycobacterial immune responses. Other groups have also found evidence of pre-existing immunity to environmental mycobacteria in healthy UK adults who are otherwise mycobacterially naive25. The leading explanation for the variable efficacy of BCG in different continents is that prior exposure to environmental mycobacteria either inhibits the replication and 'take' of BCG when this is used in subsequent vaccination, or masks the effect of BCG vaccination by providing a similar degree of antimycobacterial immunity13,26. In contrast, we find here that the immunogenicity of MVA85A appears to be enhanced by pre-existing antimycobacterial immunity induced by environmental mycobacteria.
The levels in the BCG-MVA vaccinees appear to be the highest reported T-cell responses induced by vaccination. Of particular relevance to the induction of long-lived memory immune responses, these antigen-specific T-cell responses remained significantly higher (P < 0.05) in the BCG-MVA85A group than in either the BCG or MVA85A alone group for at least 24 weeks after vaccination (Fig. 1). These data suggest that immunization with MVA85A boosts the pre-existing antimycobacterial immunity induced by BCG in these volunteers. Taken together, these results show that vaccination with MVA85A boosts pre-existing antimycobacterial immune responses induced either by environmental mycobacteria or BCG vaccination. The immunogenicity of repeated BCG immunization using these sensitive T-cell assays has not been studied and it is possible that repeated BCG vaccination could result in similar increases in T-cell responses seen after MVA85A boosting.
All of the individual peptide responses seen following vaccination with BCG and MVA85A are completely abrogated by CD4+ T-cell depletion. We have not detected any responses resulting from CD8+ T cells. This may be a technical issue that relates to the use of whole protein and peptides in the assay, rather than live BCG in a restimulation assay.
The data presented here show that MVA85A is safe and highly immunogenic, and encourage the further evaluation of this promising vaccine in tuberculosis-endemic countries. A booster MVA85A vaccination might be used either after BCG in infants or to boost immunity in teenagers before the peak of tuberculosis incidence in adolescents in developing countries. Studies are underway to evaluate the safety and immunogenicity of MVA85A in individuals latently infected with M. tuberculosis, aimed at assessing its potential utility as a postexposure vaccine. Another target group for a new tuberculosis vaccine will be HIV-infected individuals, who are at high risk of tuberculosis. Recent data on the use of MVA as an investigational therapeutic HIV vaccine suggest that MVA85A should be safe in this population27. More generally, the capacity of MVA to boost strongly pre-existing naturally evoked T-cell responses suggests potential therapeutic uses in other chronic infections.
Methods
The MVA85A vaccine.
The construction of MVA85A has previously been described5. Clinical-grade MVA85A was produced to good manufacturing practice standard by Impfstoffwerke Dessau-Tornau. A Doctors and Dentists Exemption Certificate was issued from the Medicines and Healthcare products Regulatory Agency, London, for the use of MVA85A in clinical trials.
Clinical trials.
We recruited volunteers for immunization studies under protocols approved by the Oxfordshire Research Ethics Committee and enrolled them only after obtaining written informed consent. The age range for inclusion was 18–55 and all volunteers tested seronegative for HIV, HBV and HCV at screening. We performed routine laboratory hematology and biochemistry before vaccination and all values were within normal limits. We followed all volunteers for 6 months, and took blood samples at regular timepoints. Those who received MVA85A immunizations completed a diary card recording local and systemic side effects and body temperature for 7 d following vaccination. The demographic details of the volunteers are summarized in Supplementary Table 1 online.
Vaccinations.
We conducted the first two studies in BCG-naive healthy volunteers. Those with a negative (Grade 0) Heaf test (equivalent to a tuberculin skin test of 0 mm) were vaccinated with either BCG (a single immunization with BCG Glaxo strain, 100 μl administered intradermally, n = 11) or MVA85A (5 × 107 plaque-forming units (p.f.u.) administered intradermally, 2 immunizations given 3 weeks apart, n = 14). In the third study, volunteers who had previously been vaccinated with BCG were recruited (n = 17). The median time between BCG vaccination and immunization with MVA85A was 18 years (range 0.5–38 years). We enrolled volunteers with a Heaf test not greater than grade II (equivalent to a tuberculin skin test of <15 mm) in strength in the study and immunized them with a single dose of 5 × 107 p.f.u. of MVA85A intradermally into the skin overlying the deltoid on the contralateral arm to BCG vaccination. In total, we vaccinated 31 healthy volunteers with MVA85A. Of the 14 BCG-naive volunteers, 11 received 2 immunizations, given 3 weeks apart. The remaining three received a single immunization. All of the 17 BCG-primed volunteers received a single MVA85A immunization. All volunteers completed the 6-month follow up period and there were no serious or severe adverse events in any of these studies.
Immunogenicity measures.
We used the ex vivo IFN-γ ELISPOT assay as the main immunological measure used to determine vaccine immunogenicity. We performed the assay on blood taken at the following time points: at screening before the tuberculin skin test, and then at 1, 4, 12 and 24 weeks after vaccination. These measurements were carried out on fresh PBMCs using tuberculin PPD (20 μg/ml, SSI), purified antigen 85 complex (10 μg/ml)28, and 7 pools of 9–10 15-mer peptides, overlapping by 10 amino acids (10 μg/ml final concentration of each peptide in ELISPOT well.) Briefly, we plated 300,000 PBMCs per well in 100 μl R10 (RPMI plus 10% fetal calf serum) directly onto the ELISPOT plate (MAIP S4510, Millipore) in the presence of antigen, and incubated the plate for 18 h. We used streptokinase (250 U/ml), streptodornase (12.5 U/ml) and phytohaemagglutinin (10 μg/ml) in all assays as positive controls. Assays were performed in duplicate and the results were averaged.
Epitope mapping.
Responses to individual peptides were either tested for on the first or subsequent sample after vaccination. Magnetic bead depletions (Dynal) were performed on individual peptide responses. CD4+ and CD8+ T-cell depletions were performed by 30-min incubation with monoclonal antibodies to CD4 and CD8 conjugated to ferrous beads at a ratio of five beads: one cell using M-450 (Dynal) in 200 μl R10 on ice. Antibody-coated cells were removed using a magnet (Dynal). Samples were analyzed according to undepleted and CD4+ and CD8+ T cell–depleted groups. We confirmed cell depletions by FACS scanning. Depletions were always >90% for CD8+ T cells and >97% for CD4+ T cells (data not shown).
Analysis of immunogenicity.
We analyzed the ELISPOT data by subtracting the mean number of spots in the medium and cells-alone control wells from the mean counts of spots in wells with antigens or peptide pools, and cells. We disregarded counts less than 5 spots/well. A well was considered positive if the count was at least twice that in the negative control wells and at least 5 spots more than the negative control wells. For the peptide pool wells, we summed the results across all the peptide pools for each volunteer at each timepoint. This will potentially count twice a T cell that responds to any of the 10-mer overlap regions that occur in two pools with adjacent peptides.
Statistical analysis.
We performed analysis of variance for repeated measurements using the baseline result at screening as a covariate on log-transformed data to compare between groups. We then used a Mann-Whitney test for all comparisons between groups and a Wilcoxon test for the paired comparison of screening and 24-week samples in the BCG prime-MVA85A boost group.
Cultured ELISPOT method.
For cultured ELISPOT, we stimulated 1 × 106 cryopreserved PBMC with 20 μg/ml of M. tuberculosis PPD, M. avium PPD or 10 μg/ml recombinant antigen 85A in a 24-well plate. After a 3-d incubation period at 37 °C and 5% CO2 atmosphere, we removed 500 μl of the cell culture supernatant and replaced it with 5 IU/ml Lymphocult-T (Biotest) in R10. This was repeated on day 7. On day 9 we washed the cells three times and left them to rest overnight in 37 °C and 5% CO2 atmosphere in R10. On day 10, cells were washed and resuspended in 2 ml of R10, 50 μl of cultured cells (2.5 × 104 of the initially plated cells) were transferred to duplicate wells of an ELISPOT plate and stimulated for 18 h with PPD-T 20 μg/ml, PPD-A 20 μg/ml and Ag85A 10 μg/ml. We then developed the ELISPOT plate as previously described.
Note: Supplementary information is available on the Nature Medicine website.
References
Schneider, J. et al. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat. Med. 4, 397–402 (1998).
McShane, H., Brookes, R., Gilbert, S.C. & Hill, A.V. Enhanced immunogenicity of CD4+ T-cell responses and protective efficacy of a DNA-modified vaccinia virus Ankara prime-boost vaccination regimen for murine tuberculosis. Infect. Immun. 69, 681–686 (2001).
Amara, R.R. et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292, 69–74 (2001).
Hanke, T. et al. Effective induction of simian immunodeficiency virus-specific cytotoxic T lymphocytes in macaques by using a multiepitope gene and DNA prime-modified vaccinia virus Ankara boost vaccination regimen. J. Virol. 73, 7524–7532 (1999).
McShane, H., Behboudi, S., Goonetilleke, N., Brookes, R. & Hill, A.V. Protective immunity against Mycobacterium tuberculosis induced by dendritic cells pulsed with both CD8+- and CD4+ T-cell epitopes from antigen 85A. Infect. Immun. 70, 1623–1626 (2002).
Shiver, J.W et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415, 331–335 (2002).
Gilbert, S.C. et al. Enhanced CD8 T cell immunogenicity and protective efficacy in a mouse malaria model using a recombinant adenoviral vaccine in heterologous prime-boost immunisation regimes. Vaccine 20, 1039–1045 (2002).
Colditz, G.A. et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 271, 698–702 (1994).
Rodrigues, L.C., Diwan, V.K. & Wheeler, J.G. Protective effect of BCG against tuberculous meningitis and miliary tuberculosis: a meta-analysis. Int. J. Epidemiol. 22, 1154–1158 (1993).
Randomised controlled trial of single BCG, repeated BCG, or combined BCG and killed Mycobacterium leprae vaccine for prevention of leprosy and tuberculosis in Malawi. Karonga Prevention Trial Group. Lancet 348, 17–24 (1996).
Kaufmann, S.H. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 1, 20–30 (2001).
Ellner, J.J., Hirsch, C.S. & Whalen, C.C. Correlates of protective immunity to Mycobacterium tuberculosis in humans. Clin. Infect. Dis. 30, S279–S282 (2000).
Black, G.F. et al. BCG-induced increase in interferon-gamma response to mycobacterial antigens and efficacy of BCG vaccination in Malawi and the UK: two randomised controlled studies. Lancet 359, 1393–1401 (2002).
Caruso, A.M. et al. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162, 5407–5416 (1999).
Selwyn, P.A. et al. A prospective study of the risk of tuberculosis among intravenous drug users with human immunodeficiency virus infection. N. Engl. J. Med. 320, 545–550 (1989).
van Pinxteren, L.A., Cassidy, J.P., Smedegaard, B.H., Agger, E.M. & Andersen P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur. J. Immunol. 30, 3689–3698 (2000).
McConkey, S.J. et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 9, 729–735 (2003).
D'Souza, S. et al. Mapping of murine TH1 helper T-cell epitopes of mycolyl transferases Ag85A, Ag85B, and Ag85C from Mycobacterium tuberculosis. Infect. Immun. 71, 483–493 (2003).
Launois, P. et al. T-cell-epitope mapping of the major secreted mycobacterial antigen Ag85A in tuberculosis and leprosy. Infect. Immun. 62, 3679–3687 (1994).
Huygen, K. et al. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat. Med. 2, 893–898 (1996).
Goonetilleke, N.P. et al. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J. Immunol. 171, 1602–1609 (2003).
Mwau, M. et al. A human immunodeficiency virus 1 (HIV-1) clade A vaccine in clinical trials: stimulation of HIV-specific T-cell responses by DNA and recombinant modified vaccinia virus Ankara (MVA) vaccines in humans. J. Gen. Virol. 85, 911–919 (2004).
Reece,W.H. et al. A CD4+ T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nat. Med. 10, 406–410 (2004).
Godkin, A.J., Thomas, H.C. & Openshaw, P.J. Evolution of epitope-specific memory CD4+ T cells after clearance of hepatitis C virus. J. Immunol. 169, 2210–2214 (2002).
Weir, R.E. et al. Interferon-γ and skin test responses of schoolchildren in southeast England to purified protein derivatives from Mycobacterium tuberculosis and other species of mycobacteria. Clin. Exp. Immunol. 134, 285–294 (2003).
Brandt, L. et al. Failure of the Mycobacterium bovis BCG vaccine: some species of environmental mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect. Immun. 70, 672–678 (2002).
Cosma, A. et al. Therapeutic vaccination with MVA-HIV-1 nef elicits Nef-specific T-helper cell responses in chronically HIV-1 infected individuals. Vaccine 22, 21–29 (2003).
De Bruyn, J. et al. Purification, characterization and identification of a 32 kDa protein antigen of Mycobacterium bovis BCG. Microb. Pathog. 2, 351–366 (1987).
Acknowledgements
We thank the subjects who took part in these studies. We thank I. Poulton, K. Moore and T. Berthoud for assistance and discussions, and K. Franklin for providing recombinant antigen 85A. K.H. received grants from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (G.0266.00), from Brussels Capital Region and from Damiaanaktie Belgium. This work is supported by the Wellcome Trust. H.M. is a Wellcome Trust Clinician Scientist Fellow and A.V.S.H. is a Wellcome Trust Principal Research Fellow.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A.H. is co-founder of, shareholder in and consultant to and H.M. is shareholder in and consultant to Oxxon Therapeutics PLC.
Supplementary information
Supplementary Table 1
Demographic details of subjects (PDF 6 kb)
Rights and permissions
About this article
Cite this article
McShane, H., Pathan, A., Sander, C. et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med 10, 1240–1244 (2004). https://doi.org/10.1038/nm1128
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1128
This article is cited by
-
Functional in-vitro evaluation of the non-specific effects of BCG vaccination in a randomised controlled clinical study
Scientific Reports (2022)
-
Identification of antigens presented by MHC for vaccines against tuberculosis
npj Vaccines (2020)
-
Towards new TB vaccines
Seminars in Immunopathology (2020)
-
Tuberculosis vaccine development: from classic to clinical candidates
European Journal of Clinical Microbiology & Infectious Diseases (2020)
-
Novel vaccination approaches to prevent tuberculosis in children
Pneumonia (2016)
