
Abstract
Conservation policies usually focus on in situ protection of native populations, a priority that requires accurate assessment of population status. Distinction between native and introduced status can be particularly difficult (and at the same time, is most important) for species whose natural habitat has become both rare and highly fragmented. Here, we address the status of the white elm (Ulmus laevis Pallas), a European riparian tree species whose populations have been fragmented by human activity and is protected wherever it is considered native. Small populations of this species are located in Iberia, where they are unprotected because they are considered introductions due to their rarity. However, Iberia and neighbouring regions in southwestern France have been shown to support discrete glacial refuge populations of many European trees, and the possibility remains that Iberian white elms are native relicts. We used chloroplast RFLPs and nuclear microsatellites to establish the relationship between populations in Iberia and the Central European core distribution. Bayesian approaches revealed significant spatial structure across populations. Those in Iberia and southwestern France shared alleles absent from Central Europe, and showed spatial population structure within Iberia common in recognized native taxa. Iberian populations show a demographic signature of ancient population bottlenecks, while those in Central European show a signature of recent population bottlenecks. These patterns are not consistent with historical introduction of white elm to Iberia, and instead strongly support native status, arguing for immediate implementation of conservation measures for white elm populations in Spain and contiguous areas of southern France.
Similar content being viewed by others


Introduction
Populations at the periphery of a species’ range are important in the conservation of endangered species, and of biological diversity in general (Channell and Lomolino, 2000). For many widespread tree species, it is precisely the populations at the edge of the distribution range that accumulate most of the intraspecific genetic variation, either because they correspond to glacial (micro-) refugia (for example, Kelly et al., 2010; Anderson et al., 2011), or because they represent contact zones between migrants from such refugia (Hewitt, 2004; Magri et al., 2006). Conservation of natural marginal populations of species has therefore long been considered crucial for the preservation of genetic diversity and evolutionary potential (Newton et al., 1999; Moritz, 2002). Usually, only populations thought to be native are prioritized for conservation (for example, in the Red List of the International Union for Conservation of Nature (IUCN, 2001)), and populations thought to be introduced or of dubious status are typically not protected (Decocq et al., 2004). However, it can be difficult to apply the full range of ecological, physiological and biogeographical criteria considered desirable in assessment of native status at the margins of a species’ range, raising the importance of assessing population status using genetic information.
The European white elm (Ulmus laevis Pallas) is a broadleaved, deciduous tree characteristic of river margins and damp lowland forests. It is one of the few trees tolerant to prolonged flooding (Collin, 2003), and constitutes an important foundation species in suitable habitats. Suitable riparian habitats have been widely and extensively disturbed by transformation into agricultural and pasture land, flow regulation of rivers and mining (Hooke, 2006). A combination of habitat destruction and Dutch elm disease have compromised the long-term survival of U. laevis through most of its distribution range, leaving many small and isolated populations vulnerable to genetic drift (Collin et al., 2004). The European Forest Genetic Resources programme therefore identifies U. laevis as a species requiring conservation (Collin, 2003).
The natural distribution of white elm is currently regarded as extending across central and eastern Europe, from the Ural Mountains to eastern France, and from southern Finland to the Caucasus and Bosnia (Jalas and Suominen, 1999; Collin, 2003). Populations have also long been recorded in Spain, including the Pyrenees (Lapeyrouse, 1813) and Asturias (Pastor, 1853), and were included in the first Iberian floras (Willkomm and Lange, 1861). However, on the basis of only its rarity and in the absence of any historical evidence of introduction, this species is currently considered introduced in Spain (Navarro and Castroviejo, 1993; Collin, 2003) and it has been excluded from subsequent Spanish floras (Ceballos, 1966; Ruiz de la Torre, 1971). As a result, populations of U. laevis in the Iberian Peninsula and neighbouring regions of southern France (Timbal and Collin, 1999) are unprotected. The alien status of white elm in Iberia was challenged by Segura (1973) following discovery of a previously unknown population in Central Iberia, and over the last 10 years 53 Spanish locations for this species have been found during field surveys by the Spanish Elm Conservation and Breeding Programme (SECBP). The key question is whether these geographically widespread but very local populations represent native relict fragments, or the dispersed introductions inferred for some other species (Decocq et al., 2004; Gil et al., 2004).
Fossil records, particularly pollen, can provide conclusive evidence of native status for some plants (Webb, 1985; Postigo-Mijarra et al., 2008). However, pollen is only identifiable at the genus level for European elms (Stafford, 1995). In the absence of fossil data, neutral genetic markers can offer evidence as to the native status of populations of uncertain origin. Native Iberian populations of widespread European trees are commonly divergent genetically from populations in central and eastern Europe, reflecting confinement to, and limited gene flow between, discrete glacial refugia through the Pleistocene (King and Ferris, 1998; Petit et al., 2002; Hampe et al., 2003; Rendell and Ennos, 2003; Heuertz et al., 2004; Magri et al., 2006; Maliouchenko et al., 2007). Such separation is also commonly associated with region-specific genetic variation in Iberia and immediately contiguous regions of southern France that is absent from Central Europe and regions further east.
In contrast, introduced populations are characterized by possessing a subset of genetic diversity found in source populations (Stone and Sunnucks, 1993). If introduced from a single alternative refuge population, introductions are often genetically depauperate (Webb, 1985; Stone and Sunnucks, 1993; Bossdorf et al., 2005), though introductions or colonizations from multiple populations can, together, show higher genetic diversity than any single source (for a natural example in Fagus, see Comps et al., 2000).
Under the ‘Relict hypothesis’, we expect Iberian populations to possess regionally private genetic diversity absent from Central Europe. Under the ‘Introduction hypothesis’ we expect Iberian populations to contain a subset of genetic diversity associated with the Central European core distribution of white elm, and to lack regionally private genetic variants. Here, we test these hypotheses using chloroplast (cpDNA) RFLP and nuclear (nDNA) microsatellite markers for European populations of U. laevis sampled from Spain, neighbouring regions of southern France and Central Europe. We build on a previous analysis using only samples from Europe outside Iberia (Whiteley, 2004) that identified three cpDNA haplotypes in U. laevis: a common haplotype A, extending from France to NW Russia, and two rare haplotypes—haplotype B confined to southern France and haplotype C confined to the Balkans and SW Russia. Whiteley (2004) argued that the presence of both haplotypes A and C in Russia is compatible with a core Russian glacial refuge, from which current white elm populations would have originated by range expansion. This idea was first proposed by Huntley and Birks (1983). However, Whiteley (2004) also recognizes that the southern distributions of haplotypes B and C could indicate additional refugia.
In order to further discriminate between the Relict and Introduction hypotheses, we also use a combination of analyses (Williamson-Natesan, 2005) that permit detecting population bottlenecks over recent and longer timescales. If Iberian white elms are descendants of repeated isolated introductions of small numbers of individuals from larger source populations, we expect them to show signatures of recent population bottlenecks (Garza and Williamson, 2001).
Materials and methods
Sampling
Our analysis incorporates data for 242 elms from 20 populations, 186 individuals from 11 populations in Spain and 56 individuals in 9 populations from the rest of Europe. Sampling in Iberia spanned the known distribution (Figure 1, Supplementary Table S1). Four Spanish populations (ATA, ERR, ORO and TDU) correspond to the populations considered ‘naturalized’ (but of no specified origin) in the Flora Iberica (Navarro and Castroviejo, 1993). The remainder comprises locations discovered during SECBP field surveys. Whenever possible we sampled trees separated by >50 m in order to avoid collecting leaves from closely related individuals, as this could result in a bias in allele frequency and diversity parameters estimates (Ruiz-Guajardo et al., 2010).

Location of the 20 populations of U. laevis included in this study (white circles). Population codes correspond to Table 1. Spanish populations were chosen to cover the known species range. The shaded area shows the most recent distribution map of U. laevis published by the European Forest Genetic Resources Programme (EUFORGEN), including the populations of South France (Collin, 2003). (Map created using Lambert’s equal-area projection).
The 56 European U. laevis DNA samples from France, Belgium and Germany were obtained from the Royal Botanic Garden of Edinburgh (RBGE) (see Figure 1). These are a subset of samples from the EU project ‘Coordination for conservation, characterization, collection and utilization of genetic resources of European Elms’ (RESGEN CT96-78) (Collin et al., 2004) and were included to represent genetic diversity in the core distribution for this species (shaded grey in Figure 1).
DNA extraction and scoring of molecular markers
We extracted DNA from fresh leaves using a modified protocol from the study by Dumolin et al. (1995), in which ∼1 cm2 of ground leaf material and 20–30 mg of insoluble polyvinylpirrolidone were combined with 1 ml of 1 × ATMAB buffer, plus 0.1% w/v 1,4-Diothiothreitol.
Chloroplast DNA (cpDNA) was analysed by PCR–RFLP using regions trnS–trnfM, trnH–trnK and trnD–trnT (Demesure et al., 1995), previously shown to vary in European elms (Gil et al., 2004; Whiteley, 2004). Amplicons were digested with restriction enzymes (fragment-enzyme combinations: SfM-MvaI, HK-HaeIII, DT-TaqI). Restriction fragments were separated by electrophoresis in 8% non-denaturating polyacrylamide gels and visualized using silver staining. Chloroplast haplotypes were then assigned by combining the observed polymorphisms of each PCR fragment-restriction enzyme combination. Correspondence of observed haplotypes to those previously described (Whiteley, 2004) was established with reference to 39 common samples stored at the RBGE (Whiteley, pers. comm.).
To verify sequence conservation of the PCR–RFLP haplotypes across their distribution ranges, the two observed haplotypes for the SfM digest region (the only variable region) were sequenced for eight samples (four from Spain, two from France, one from Belgium and one from Germany, specified in Supplementary Table S1), spanning the distribution of both haplotypes. SfM products were sequenced on a CEQ 8000 Beckman-Coulter automated sequencer (Beckman-Coulter, Brea, CA, USA), and edited and aligned using Geneious Pro 5.1.7 (Biomatters, Auckland, New Zealand).
Five nuclear microsatellite loci (ulm2, ulm3, ulm6, ulm9 and ulm19) developed for U. laevis were screened following PCR conditions given in Whiteley et al. (2003). PCR products were denatured and separated in 6% (w/v) acrylamide:bisacrylamide (19:1) gels using a Li-Cor 4300 automatic sequencer (Li-Cor Biosciences, Lincoln, NE, USA), and alleles were scored using Gene ImagIR 3.56 (Scanalytics, Milwaukee, WI, USA). A molecular size standard (Sequamark, Invitrogen, Carlsbad, CA, USA) was included in each gel for the identification of allele sizes. All 242 samples were screened for all loci.
Bayesian identification of genotype pools in U. laevis
Two Bayesian clustering programs were used to assess population structure in the microsatellite data set: STRUCTURE v.2.1 (Pritchard et al., 2000) and BAPS v.4.0 (Corander et al., 2008). First, we used STRUCTURE to reveal individual-based patterns in genetic variation without prior information on sample geographic origin. We performed three independent runs for each value of K from 1–21 (number of sampling locations plus one). The no admixture ancestry model was assumed, as it is more successful at detecting subtle differentiation (Pritchard and Wen, 2003). We used a MCMC length of 1 200 000 with burnin length of 200 000 (enough for the summary statistics to have converged). For values of K ⩾2 the proportion of each individual’s genotype assigned to each genetic cluster was roughly symmetric, indicating no population structure (Pritchard and Wen, 2003). To detect subdivision within the two clusters observed, the analysis was repeated independently for individuals included within them (Pritchard and Wen, 2003). Second, we used BAPS to incorporate sample geographic information. The 56 samples from the RBGE represent individuals sampled over larger geographical areas rather than from discrete populations. To allow incorporation of geographic location information for these samples, we divided them into nine groups based on geographic proximity and shared watershed (Supplementary Table S1). Geographic coordinates of each group were approximated using the geographic centroid of the individual sample origins. These nine groups were added to the 11 Iberian populations to give 20 geographic groupings in the BAPS analysis using the ‘clustering of groups of individuals’ option. There was not enough information in the data set to get conclusive results using individual clustering. We carried out 10 independent simulations for each value of K from 1–21.
Measures of genetic diversity and population structure
We compared the diversity and distribution of genetic variation for both cpDNA and microsatellite markers for each of the 11 Spanish populations, and for the genotype groupings supported by BAPS. We did not consider the nine post hoc Central European sample sets as populations in this context. Using the distance between observed haplotypes, cpDNA genetic diversity parameters corrected for variation in population sizes (Pons and Petit, 1995) were calculated for each population (hk), and region (hS), and overall populations (hT), together with the coefficient of genetic differentiation among subpopulations (GST) within Iberia and among BAPS clusters using HAPSTEP (http://www.pierroton.inra.fr/genetics/labo/Software/; Pons and Petit, 1996). An analysis of molecular variance (AMOVA) was run using GENALEX (Peakall and Smouse, 2006) to test significance of genetic diversity partitioning among BAPS clusters, among groups of individuals within BAPS clusters, and within populations, based on 999 permutations.
From the microsatellite data, we estimated mean number of alleles (A), mean allelic richness per sample (Rs), the number of regionally private alleles (Ap), percentage of polymorphic loci (P), mean expected (HE) and observed (HO) heterozygosities and inbreeding coefficients (FIS) using FSTAT (Goudet, 2001). We calculated these parameters for the Spanish populations and, at the regional level, for the BAPS groups. The latter regional analysis is also intended to reduce potential biases introduced by small sample sizes in the Central European populations. We tested the statistical significance of differences in diversity parameters between regions in FSTAT using either a Mann–Whitney U-test (A) or permutation tests (Rs, HE and FIS). We tested departures from Hardy-Weinberg equilibrium (HWE) within populations, within regions and overall using permutation tests in FSTAT. We estimated the population differentiation parameter FST (Weir and Cockerham, 1984) for populations within Iberia, and between BAPS groups using FSTAT; significant deviations from zero population differentiation were assessed with permutation tests. We carried out an AMOVA analysis of microsatellite diversity using GENALEX at the same hierarchical levels used in analysis of the cpDNA data.
Finally, we used BayesAss (Wilson and Rannala, 2003) to estimate rates of recent migration (over the last several generations, which correspond to the time scale of putative human-mediated movement of U. laevis) among BAPS groups. We performed the analysis with 30 000 000 iterations, with a burnin period of 999 999, and a sampling frequency of 2000. Delta values for allele’s frequencies, migration rate and inbreeding were set to 0.15 (default value).
Demographic analysis
We used M-value (Garza and Williamson, 2001) and BOTTLENECK v.1.2.02 (Cornuet and Luikart, 1996) analysis of microsatellite data to assess the evidence for population bottlenecks in all Iberian populations containing >11 individuals, and, across western Europe, in the genetic clusters supported by BAPS. M-value is the mean ratio of the number of alleles to the range in allele size. This ratio decreases when a population is reduced in size, and M-values of <0.68 are indicative of a genetic bottleneck (Garza and Williamson, 2001). For the BOTTLENECK analysis, we ran a Wilcoxon sign rank test with 10 000 iterations under the infinite alleles (IAM), stepwise mutation (SMM) and the two-phase (TPM) models. The SMM and IAM represent two extreme mutation models, with the SMM being the most conservative model to test for heterozygosity excess (Cornuet and Luikart, 1996). As the TPM may more closely capture the mutation behaviour of microsatellites (Di Rienzo et al., 1994; Williamson-Natesan, 2005), here we emphasize results with this approach. Simulation studies (Williamson-Natesan, 2005) have shown that tests based on the range in allele size conditioned on the number of alleles (M-value) are better able to detect a bottleneck lasting several generations followed by demographic recovery (see also Garza and Williamson, 2001). In contrast, tests based on heterozygosity given the number of alleles (BOTTLENECK) are better able to identify recent, less-severe bottlenecks. Our premise is that because Iberian populations have been rare throughout recorded history, a bottleneck signature in allele-size distributions is more likely to be associated with remnant native populations than a recent introduction.
Results
Bayesian analysis of population structure
STRUCTURE analysis supported division of Europe-wide samples into two clusters (named I and II) that correspond to two geographic regions (Table 1). Cluster I comprised six populations in southwestern Europe: five in western and southern Spain (CNA, AME, BLU, JVE and PSG), all with a high (>0.750) proportion of membership assigned to cluster I, and the southern-most French group of individuals (GAR), with an assignment probability of 0.649. The remaining Iberian populations and European groups were assigned to Cluster II with probabilities ranging from 0.537–0.995. When each cluster was analysed independently, only cluster II showed evidence of further substructure, and was divided into two subclusters (IIa and IIb, Table 1). Subcluster IIa is formed by five populations from central and northern Spain (PER, ATA, ERR, ORO and MEN) plus a group of individuals from southern France (VIE). Subcluster IIb comprises the groups from the core distribution of the species in central Europe (SAO, SEI, NFB, URH, UDA, MRH and NGE), plus one Spanish population (TDU, with only two individuals). STRUCTURE analysis is a conservative grouping approach agnostic of prior geographic origin. Grouping of TDU in subcluster IIb is the only geographic inconsistency observed, suggesting that it is valid to use BAPS to incorporate further geographic information.
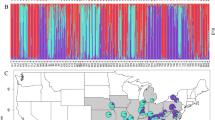
BAPS results provided strong posterior probability (88%) support for allocation of individuals into five genetic groups showing clear geographical structure (Figure 2). Cluster A includes the core Central European populations from northern and eastern France, Belgium and Germany, whereas clusters B–E comprise populations in Spain, western and southern France. Cluster B comprises 5 populations in central and northern Spain and the two southern-most French populations. This BAPS cluster contains most of the same individuals in STRUCTURE’s subcluster IIa, with the addition of the individuals from site TDU in central Spain (the outlier in STRUCTURE subcluster IIb) and GAR (a population assigned with relatively low probability to in STRUCTURE cluster I). Clusters C and E each consisted of one Spanish population (MEN and CNA respectively). Cluster D comprises four Spanish populations located in separate watersheds (AME, BLU, JVE and PSG), also grouped together in STRUCTURE cluster I. It is worth noticing how the incorporation of geographical information in the BAPS analysis seems to help detecting more detailed genetic grouping than STRUCTURE.

Distribution of cpDNA haplotypes and clusters of populations based on five nuclear microsatellites for U. laevis. The two cpDNA haplotypes are shown in blue (A) and red (B). Pie charts represent the proportion of individuals observed for each haplotype in each population (Table 1), with sizes proportional to the number of individuals analysed. For the nuclear SSR markers, the map shows the Voronoi tessellation and assignment to the five clusters (A–E) obtained with BAPS (map created using Lambert’s equal-area projection).
Genetic diversity
Chloroplast DNA
Of the three cpDNA regions analysed by PCR–RFLP, only fragment SfM was polymorphic, with two size variants in one of the two fragments obtained after digestion with MvaI. This polymorphism distinguished two cpDNA haplotypes—A (1241 base pairs, bp) and B (1248 bp) differing by a 7-bp sequence (TAATAAA) present in haplotype B. Haplotype A was the commonest haplotype (75.3% of 150 samples analysed) (Table 2, Figure 2). It was the only variant detected in Central European samples, and was also found in nine Spanish populations (of which it was fixed in seven; Table 2). Haplotype B (24.7% of individuals) was restricted to Iberia, being found in four Spanish populations, and fixed in two (ERR, ORO) (Table 2). Iberian populations showed low within-population diversity (hS=0.067). However, the presence of haplotype B in these populations resulted in moderate total diversity (hT=0.440) and high population differentiation (GST=0.847). When BAPS groupings were considered, all cpDNA variation was restricted to cluster B in central-north Iberia plus southern France (hk=0.440), with all other clusters fixed for haplotype A, resulting in lower total genetic diversity (hT=0.274). Genetic differentiation among clusters was relatively high (GST=0.679), although lower than among Iberian populations alone. AMOVA for Iberian populations showed that the majority (52%) of the genetic variation was found among BAPS clusters, with ∼35% of variation observed among populations within clusters and only 13% within clusters (Supplementary Table S2). These percentages represent significant (<0.001) spatial structure at each level.
Nuclear microsatellites
Most loci were polymorphic in all 11 Iberian populations, except for 3 of them (AME, JVE and PSG) that were monomorphic at one locus (Table 2). Overall allelic diversity of Iberian populations was relatively low (A=2.62, Rs=1.91). In the Spanish populations the lowest values for most genetic diversity parameters were observed in AME (A=2.2, Rs=1.59), and the highest in PER (A=3.2) and TDU (Rs=2.40) (Table 2). Allele frequencies for all groupings were in Hardy–Weinberg equilibrium.
Microsatellite data revealed significant population structure among Iberian populations (FST=0.155; P<0.001) and across all groups of individuals (global FST=0.143; P<0.001). At the regional level, the test for population differentiation among BAPS clusters was highly significant (FST=0.126; P<0.001; Supplementary Table S3). Cluster A (Central Europe) showed the highest values in genetic diversity parameters, in terms of mean number of alleles per locus (5.0), mean allelic richness per sample (3.87) and gene diversity (HE=0.513). Three BAPS clusters showed private alleles: cluster A (with eight private alleles) in Central Europe, and clusters B and D in SW Europe (with one private allele each). The AMOVA among BAPS clusters also showed significant (P<0.001) differentiation at all levels of partitioning (Supplementary Table S2): most (71%) of the variation was found within populations, with 19% among BAPS clusters and only 10% among populations within clusters.
Migration rates among BAPS clusters
Only in two pair-wise comparisons between BAPS clusters were BayesAss migration rates greater than 10%. These were migration from cluster B into cluster A (m=0.21, s.e.=0.04) and migration from cluster D into cluster E (m=0.12, s.e.=0.10) (Supplementary Table S4). The observed migration rate (and 95% confidence interval) in simulated data sets of five populations for when there is no information in the data is 0.0417 (1.79 × 10−5, 0.185). Assuming normality in the distribution of migration rates, only the first migration rate (from cluster B into cluster A) resulted to be significantly different from the no-information migration rate (t-test P=0.001).
Demographic analysis
The results of the bottleneck tests varied with the method used (Table 3). Analysis of allele size distributions (M-values) identified bottlenecks in six out of the eight Iberian populations tested (M-value<0.68), and did not detect a bottleneck signal in the two European ones (URH and MRH; Table 3). All BAPS clusters from SW Europe (B, C, D and E) showed a signal of bottlenecking with M-value.
Analyses of heterozygosity (BOTTLENECK) showed significant heterozygosity excess (P<0.05, Wilcoxon test), indicative of population bottlenecking for five populations under the two-phase model (Table 3), comprising four Spanish populations (ATA, CNA, ERR and PER). Only BAPS clusters B and E showed evidence of bottlenecking by this test. No population or genetic cluster showed a heterozygosity deficit indicative of population expansion.
Discussion
Is U. laevis native to the Iberian Peninsula?
Our analysis substantially extends the range sampled in previous work on U. laevis (Whiteley, 2004; Nielsen and Kjær, 2010). We show that though populations in Iberia have low absolute genetic diversity, they contain regionally private genetic variation in both chloroplast sequence (haplotype B) and nuclear microsatellites. Differentiation between Iberia/SW France and Central Europe, and spatial structure among Iberian populations are further supported by regional differentiation of BAPS genotype clusters.
Two general explanations exist for this pattern. The first is that Iberian populations of U. laevis represent previously unrecognized native populations. Many widespread temperate trees in the Western Palaearctic show strong longitudinal population structure associated with discrete putative glacial refugia (for example, Petit et al., 2002; Hampe et al., 2003; Magri et al., 2010). In all cases so far studied, each refuge is characterized by private genetic variation, accumulated during glacial advance. In many tree species, and as we find for U. laevis, genetic variants diagnostic of native Iberian populations also extend into southern France, particularly in the west (Petit et al., 2002; Magri et al., 2010). Iberian populations do not show a simple subset of genetic diversity present in Central Europe, a pattern expected if Iberian populations are recently derived by introduction from this source (Stone and Sunnucks, 1993). Furthermore, the only significant recent migration detected is from southwestern Europe into Central Europe, and not the other way around. Unless we posit that the Iberia-specific genetic variation evolved rapidly since an introduction (and this is unlikely due to the low mutation rate of cpDNA (Clegg et al., 1994)), the genetic diversity observed in Iberian populations is not compatible with introduction from Central Europe.
A second possible explanation is that Iberian populations are the result of one or more introductions from alternative, genetically divergent, sources within the native range of U. laevis (Figure 1) that harbour genotypes differing from those in Central Europe. In contrast to many European trees, U. laevis is absent from Italy, which can thus be discounted as a possible source. White elm is native to the Balkans, which could thus represent a possible source. However, previous work by Whiteley (2004) indicates that this region is occupied by a different haplotype, C, also present in Russia. In the absence of an Italian refuge for U. laevis, the Balkans are also the most likely refugial origin of the Central European populations sampled in our study and fixed for chloroplast haplotype A. Both lines of reasoning argue against a Balkan origin for Iberian populations, but cannot absolutely exclude this possibility. Further sampling of Balkan populations is required to test this hypothesis.
Regardless of their origin, demographic analyses also argue against recent introduction of Iberian populations. Populations and BAPS clusters from Spain and southwestern France show a more consistent bottleneck signature in allele-size distributions than in heterozygosity excess. This in turn suggests that underlying demographic effects lasted for several generations, rather than being recent phenomena (Garza and Williamson, 2001; Williamson-Natesan, 2005). It is possible for introduced populations to carry with them demographic signatures from their source population(s)—but only where the introduction comprises relatively large numbers of individuals (for example, Stone et al., 2007). There is no historical evidence for any large-scale transport of white elm (in contrast, for example, to Field elm Ulmus minor; Gil et al., 2004). We suggest that the bottleneck signatures in Iberian populations reflect natural population fragmentation associated with Holocene expansion of more xeric habitats in Iberia, rather than impacts of recent introduction for gardening as suggested by Navarro and Castroviejo (1993), and for which no documentary evidence exists. Associated genetic drift could explain the lower allelic richness observed in southwestern Europe compared with Central Europe, where more suitable climatic conditions promote larger population sizes. In contrast, recent bottlenecks detected by heterozygosity excess in half of the Spanish populations, in Central Europe (URH) and in the Netherlands (Nielsen and Kjaer, 2010) may have resulted from intense and historically recent anthropogenic landscape transformation (Valbuena-Carabaña et al., 2010).
Palaeobotanic and modelling evidence are also congruent with the native status hypothesis of U. laevis. Palaeobotanical records show the presence of Ulmus in the Iberian Peninsula since the Miocene and throughout the Pleistocene and Holocene (López, 2000), proving that Iberia was a glacial refuge for Ulmus. Unfortunately, U. minor, U. glabra and U. laevis wood (Schweingrüber, 1990) and pollen (Stafford, 1994) cannot be distinguished. Fossil macro-remains such as fruits or leaves discriminate between elm species, but unfortunately these are very rare (García-Amorena et al., 2008). Iberia has also been determined to be a glacial refugia for the three elm species by climatic modelling (Svenning et al., 2008). The combination of palaeoecology, phylogeography and species distribution modelling have a great potential to inform each other, as they provide complementary information, and have helped interpret glacial refuge for several tree species in Iberia (Rodfíguez-Sánchez et al., 2010).
Assuming that Iberia acted as a glacial refugium, the phylogeographic pattern observed in U. laevis is similar to those observed in other widely distributed, non-cultivated, European tree taxa (that is, Petit et al., 2002; Magri et al., 2006). Before U. laevis, other tree taxa previously considered introduced in the Iberian peninsula have been proven to be native by the use of genetic markers, like chestnut (Fineschi et al., 2000), olive (Breton et al., 2006), stone pine (Vendramin et al., 2008). The latter, like U. laevis, showed very little genetic diversity across its broad Mediterranean range. Although in these species ancient cultivation by humans may have justified some doubts about their native status, this has never been the case for U. laevis, which has been considered introduced in the absence of any historical evidence solely due to its rarity in Iberia. In contrast, the genetic data presented in this study provide evidence that U. laevis populations in SW Europe are more likely to be native.
Regional variation in population structure
Our results (FST, AMOVA and BAPS clusters) show significant genetic differentiation within Iberia, and between Central and SW Europe, but genetic homogeneity within Central Europe. Similarly, Whiteley (2004) found no differentiation among five Central and North Eastern European populations of U. laevis, but significant differentiation between these populations and each of southwestern France and Finland. Genetic differentiation is frequently higher within natural than introduced populations (Bossdorf et al., 2005), but reduced gene flow between recently established populations can also cause genetic subdivision where no differentiation is found in the centre of a species’ range (Stone and Sunnucks, 1993). Population substructure alone is thus agnostic on the introduced versus native status of U. laevis. However, if Iberian populations are considered native, differentiation among them may be significant for conservation and management of regional diversity.
Whatever its cause, the small size of Iberian populations of U. laevis together with self-incompatibility make this species particularly susceptible to population bottlenecks and associated genetic drift (Nielsen and Kjaer, 2010). The greatest risk faced by U. laevis in Iberia is the on-going fragmentation and small size of its populations. In Spain, we only found U. laevis in 53 locations, 21 (40%) comprising just one or two isolated individuals. Similarly, in South France U. laevis appears in groups of trees or scattered individuals found in just nine sites (Timbal and Collin, 1999). Human disruption of riparian habitats, by agriculture, sand and gravel mining, hydrological control of rivers and urbanization, are the greatest threats to these U. laevis populations (Collin, 2003). An example of the latter is the population of Palazuelos de Eresma (PER). This stand has lost 22% of its mature elms (28 of 130) in the last 5 years while being transformed into a golf course and urban complex. Tardajos de Duero (TDU) has also experienced severe recent population size reduction, from several individuals (Segura, 1973) to just two trees when sampled in 2005.
In Spain and South France, white elm populations occupy a total area of <10 km2. These severely fragmented populations also show a continuous decline in area, quality of habitat and number of mature individuals. Following the IUCN Red List criteria (IUCN, 2001), if U. laevis is considered native in Iberia it should be considered as critically endangered (B2ab(iii+v)). However, as a presumed alien species in Iberia, U. laevis currently remains excluded from the Red List of Spanish Vascular Flora (VVAA, 2000). Within the European Union, the Habitat and Species Directive (Council Directive 92/43/EEC) established the list of natural habitat types of community interest for which special areas of conservation (Natura 2000 network) have been designated since 1992. Although habitat 91F0 (Section 91) of the Directive set up the basic framework for the conservation of populations of U. laevis in Central and Eastern Europe, none of the prioritized habitats listed for Mediterranean deciduous forests (Section 92, Annex I) supports conservation of U. laevis in the Iberian Peninsula and southwestern France. Inclusion of a species in these lists is a de facto prerequisite for direct action towards population protection or restoration (Mace, 2004). We argue that the balance of evidence now supports inclusion of U. laevis in the Iberian Red List, and initiation of associated regional conservation measures. At the habitat level, native status for Iberian white elm would strengthen the argument for inclusion of Mediterranean riparian forests with U. laevis under Section 92 of the Directive. Unless effective action is taken immediately, the issue will become moot because there will be no Iberian populations left to conserve.
We propose the definition of evolutionary significant units, and of management units within these (Moritz, 2002), for U. laevis conservation (Supplementary Information). The wide range of in situ and ex situ conservation measures suggested for U. laevis by Collin et al. (2004) should be based on the proposed management units. Efforts should focus on augmentation of remnant populations and, where possible, introduction to suitable riparian environments to facilitate connectivity among existing populations. Populations with very low numbers of adult trees (that is, PSG and TDU) could be increased with seedlings from larger populations within the same management unit to minimize risks of inbreeding depression (Moritz, 1999) and maintaining their evolutionary potential.
Although susceptible to Dutch elm disease, U. laevis is far less attractive to elm bark beetles transmitting Dutch elm disease than U. minor (Webber, 2000), and in many Central European rivers has replaced the latter as the most common elm species following Dutch elm disease pandemics (although with much lower population sizes) (Mackenthum, 2004). As long as the insect vectors do not change their feeding preferences, the genetic resources of U. laevis are not really endangered by Dutch elm disease. After the great losses suffered by U. minor in the last decades, and with other riparian trees currently declining due to other fungal diseases (i.e., alder, Alnus glutinosa; Solla et al., 2010), conservation and promotion of U. laevis can be very valuable for the restoration and preservation of SW European riparian ecosystems.
Conclusions
Our data shows that Iberian populations of U. laevis, traditionally considered non-native and of no conservation interest, are genetically distinct from populations in Central Europe and may represent relict populations of an Iberian glacial refuge. This phylogeographic pattern would be in accordance with those observed in other European tree taxa (Petit et al., 2002; Magri et al., 2006). Therefore, attending to our genetic data, and to historic and palaeobotanic evidence, we consider it is much more likely that Iberian populations of U. laevis are native rather than introduced. Should this native status be confirmed by a larger scale study, it would represent a rare addition of a new tree species to Flora ibérica, and it would also corroborate native status for U. laevis in South France, as proposed by Timbal and Collin (1999). Nevertheless, these populations require immediate and appropriate management measures for the protection of existing individuals and of the species genetic resources they represent.
Data archiving
Chloroplast sequences were deposited in GenBank: accession numbers KF515559–KF515566. Genotype data were deposited in the Dryad repository: doi:10.5061/dryad.2r6m4.
References
Anderson LL, Hu FS, Paige KN . (2011). Phylogeographic history of white spruce during the last glacial maximum: uncovering cryptic refugia. J Hered 102: 207–216.
Bossdorf O, Auge H, Lafuma L, Rogers W, Siemann E, Prati D . (2005). Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia 144: 1–11.
Breton C, Tersac M, Bervillé A . (2006). Genetic diversity and gene flow between the wild olive (oleaster, Olea europaea L.) and the olive: several Plio-Pleistocene refuge zones in the Mediterranean basin suggested by simple sequence repeats analysis. J Biogeogr 33: 1916–1928.
Ceballos L . (1966) Mapa forestal de España, escala 1:400.000. Dirección General de Montes, Caza y Pesca Fluvial: Madrid, Spain.
Channell R, Lomolino MV . (2000). Dynamic biogeography and conservation of endangered species. Nature 403: 84–86.
Clegg MT, Gaut BS, Learn GH, Morton BR . (1994). Rates and patterns of chloroplast DNA evolution. Proc Natl Acad Sci USA 91: 6795–6801.
Collin E . (2003) EUFORGEN Technical Guidelines for genetic conservation and use for European white elm (Ulmus laevis). International Plant Genetic Resources Institute (IPGRI): Rome, Italy.
Collin E, Rusanen M, Ackzell L, Bohnens J, Aguiar DA, Diamandis S et al. (2004). Methods and progress in the conservation of elm genetic resources in Europe. Invest Agra-Sist R 13: 261–272.
Comps B, Gomory D, Letouzey J, Thiébaut B, Petit RJ . (2000). Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157: 389–397.
Corander J, Sirén J, Arjas E . (2008). Bayesian spatial modeling of genetic population structure. Computation Stat 23: 111–129.
Cornuet JM, Luikart G . (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB . (1994). Mutational processes of simple-sequence repeat loci in human populations. PNAS 91: 3166–3170.
Decocq G, Bordier D, Wattez JR, Racinet P . (2004). A practical approach to assess the native status of a rare plant species: the controverse of Buxus sempervirens L. in northern France revisited. Plant Ecol 173: 139–151.
Demesure B, Sodzi N, Petit RJ . (1995). A set of universal primers for amplification of polymorphic noncoding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4: 129–131.
Dumolin S, Demesure B, Petit RJ . (1995). Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91: 1253–1256.
Fineschi S, Taurchini D, Villani F, Vendramin GG . (2000). Chloroplast DNA polymorphism reveals little geographical structure in Castanea sativa Mill. (Fagaceae) throughout southern European countries. Mol Ecol 9: 1495–1503.
García-Amorena I, Morla C, Rubiales JM, Gómez-Manzaneque F . (2008). Taxonomic composition of the Holocene forests or the northern coast of Spain, as determined from their macroremains. Holocene 18: 819–829.
Garza JC, Williamson EG . (2001). Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318.
Gil L, Fuentes-Utrilla P, Soto A, Cervera MT, Collada C . (2004). English elm is a 2,000-year-old Roman clone. Nature 431: 1053–1053.
Goudet J . (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3) Available from http://www2.unil.ch/popgen/softwares/fstat.htm last succesful access March 2007). Updated from: Goudet J, 1995. FSTAT (vers. 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Hampe A, Arroyo J, Jordano P, Petit RJ . (2003). Rangewide phylogeography of a bird-dispersed Eurasian shrub: contrasting Mediterranean and temperate glacial refugia. Mol Ecol 12: 3415–3426.
Heuertz M, Fineschi S, Anzidei M, Pastoreli R, Salvini D, Paule L et al. (2004). Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13: 3437–3452.
Hewitt GM . (2004). Genetic consequences of climatic oscillations in the Quaternary. Phil Trans R Soc B 359: 183–195.
Hooke JM . (2006). Human impacts on fluvial systems in the Mediterranean region. Geomorphology 79: 311–335.
Huntley B, Birks HJB . (1983) An Atlas of Past and Present Pollen Maps for Europe: 0-13,000 Years Ago. Cambridge University Press: Cambridge, UK.
IUCN. (2001) IUCN Red List Categories and Criteria: Version 3.1. IUCN Species Survival Commission: Gland – Cambridge.
Jalas J, Suominen J . (1999). Ulmus laevis (Ulmaceae). Atlas Florae Europaeae Database. Finnish Museum of Natural History available at, http://www.fmnh.helsinki.fi/english/botany/afe/ Last access: August 2007.
Kelly A, Charman DJ, Newnham RM . (2010). A Last Glacial Maximum pollen record from Bodmin Moor showing a possible cryptic northern refugium in southwest England. J Quat Sci 25: 296–308.
King RA, Ferris C . (1998). Chloroplast DNA phylogeography of Alnus glutinosa (L.) Gaertn. Mol Ecol 7: 1151–1161.
Lapeyrouse P . (1813) Histoire abrégée des plantes des Pyrénées et ininéraire des botanistes dans ces montagnes. L’Imprimerie de Bellegarrigue: Toulouse.
López RA . (2000). Paleobotánica de los olmos. In: Gil L, Solla A, Iglesias S (eds) Los olmos ibéricos Conservación y mejora frente a la grafiosis, Organismo Autónomo Parques Nacionales: Madrid pp 49–68.
Mace GM . (2004). The role of taxonomy in species conservation. Phil Trans R Soc B 359: 711–719.
Mackenthum GL . (2004). The role of Ulmus laevis in German floodplain landscapes. Invest Agra-Sist R 13: 55–63.
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gomory DSA et al. (2006). A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171: 199–221.
Maliouchenko O, Palmé AE, Bounamici A, Vendramin GG, Lascoux M . (2007). Comparative phylogeography and population structure of European Betula species, with particular focus on B. pendula and B. pubescens. J Biogeogr 34: 1601–1610.
Moritz C . (1999). Conservation units and translocations: strategies for conserving evolutionary processes. Hereditas 130: 217–228.
Moritz C . (2002). Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst Biol 51: 238–254.
Navarro C, Castroviejo S . (1993). Ulmus. In: Castroviejo, et al (eds) Flora Iberica Vol. III Plumbaginaceae (partim)-Capparaceae, Real Jardín Botánico. CSIC: Madrid. pp 244–248.
Newton AC, Allnutt TR, Gillies ACM, Lowe AJ, Ennos RA . (1999). Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol 14: 140–145.
Nielsen LR, Kjær ED . (2010). Fine-scale gene flow and genetic structure in a relic Ulmus laevis population at its northern range. Tree Genet Genomes 6: 643–649.
Pastor P . (1853). Diagnóstico agrícola sobre la provincia de Asturias. Memoria de la Real Academia de Ciencias de Madrid 1, (parte 3) Madrid, Spains.
Peakall R, Smouse PE . (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Petit RJ, Csaikl UM, Bordacs S, Burg K, Coart E, Cottrell J et al. (2002). Chloroplast DNA variation in European white oaks: phylogeography and patterns of diversity based on data from over 2600 populations. Forest Ecol Manag 156: 5–26.
Pons O, Petit RJ . (1995). Estimation, variance and optimal sampling of gene diversity. Theor Appl Genet 90: 462–470.
Pons O, Petit RJ . (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144: 1237–1245.
Postigo-Mijarra JM, Gómez-Manzaneque F, Morla C . (2008). Survivial and long-term maintanence of tertiary trees in the Iberian Peninsula during the Pleistocene: first record of Aesculus L. (Hippocastanaceae) in Spain. Veg Hist Archaebot 17: 351–364.
Pritchard JK, Wen W . (2003). Documentation for structure software: Version 2. Department of Human Genetics, University of Chicago (USA). Available at, http://pritch.bsd.uchicago.edu/software/structure2_1.html (last visit Nov 2011)..
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rendell S, Ennos RA . (2003). Chloroplast DNA diversity of the dioecious European tree Ilex auifolium L. (English holly). Mol Ecol 12: 2681–2688.
Rodfíguez-Sánchez F, Hampe A, Jordano P, Arroyo J . (2010). Past tree range dynamics in the Iberian Peninsula inferred through phylogeography and plaeodistribution modelling: A review. Rev Palaeobot Palyno 162: 507–521.
Ruiz de la Torre J . (1971) Árboles y arbustos de la España Peninsular 1st edn. ETSI de Montes: Madrid, Spain.
Ruiz-Guajardo JC, Schnabel A, Ennos RA, Preuss S, Otero-Arnaiz A, Stone GN . (2010). Landscape genetics of the key African acacia species Senegalia mellifera (Vahl)—the importance of the Kenyan Rift Valley. Mol Ecol 19: 5126–5139.
Schweingrüber FH . (1990). Anatomy of European. Woods., Ed Swiss Federal Institute for Forest, Snow and Landscape Research,Bern.
Segura A . (1973). De la flora soriana y circumsoriana. Pirineos 109: 35–49.
Solla A, Pérez-Sierra A, Corcobado T, Haque MM, Diez JJ, Jung T . (2010). Phytophthora alni on Alnus glutinosa reported for the first time in Spain. Plant Pathology 59: 798.
Stafford PJ . (1995). The Northwest European pollen flora. Ulmaceae. Rev Palaeobot Palyno 88: 25–46.
Stone GN, Challis RJ, Atkinson RJ, Csóka G, Hayward A, Mutun S et al. (2007). The phylogeographic clade trade: tracing the impact of human-mediated dispersal on the colonisation of northern Europe by the oak gallwasp Andricus kollari. Mol Ecol 16: 2768–2781.
Stone GN, Sunnucks P . (1993). Genetic consequences of an invasion through a patchy environment—the cynipid gallwasp Andricus quercuscalicis (Hymenoptera: Cynipidae). Mol Ecol 2: 251–268.
Svenning JC, Normand S, Kageyama M . (2008). Glacial refugia of temperate trees in Europe: insights from species distribution modelling. J Ecol 96: 1117–1127.
Timbal J, Collin E . (1999). L’orme lisse (Ulmus laevis Pallas) dans le Sud de la France: répartition et stratégie de conservation des ressources génétiques. Revue Forestière Française LI: 593–604.
Valbuena-Carabaña M, López de Heredia U, Fuentes-Utrilla P, González-Doncel I, Gil L . (2010). Historical and recent changes in the Spanish forests: A socio-economic process. Rev Palaeobot Palyno 162: 492–506.
Vendramin GG, Fady B, González-Martínez SC, Hu FS, Scotti I, Sebastiani F et al. (2008). Genetically depauperate but widespread: the case of an emblematic Mediterranean pine. Evolution 62: 680–688.
VVAA. (2000). Lista Roja de Flora Vascular Española (valoración según categorías UICN). Conservación vegetal 6: 11–38.
Webb DA . (1985). What are the criteria for presuming native status? Watsonia 15: 231–236.
Webber JF . (2000). Insect vector behavior and the evolution of Dutch elm disease. In: Dunn CP (ed) The elms: breeding, conservation, and disease management. Kluwer Academic Publishers: Boston. pp 47–60.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whiteley RE . (2004) Quantitative and molecular genetic variation in Ulmus laevis Pall PhD Thesis Swedish University of Agricultural Sciences.
Whiteley RE, Black-Samuelsson S, Clapham D . (2003). Development of microsatellite markers for the European white elm (Ulmus laevis Pall.) and cross-species amplification within the genus Ulmus. Mol Ecol Notes 3: 598–600.
Williamson-Natesan EG . (2005). Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6: 551–562.
Willkomm HM, Lange JMC . (1861) Prodromus florae hispanicae I, Stuttgart. Pp 248–249.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Acknowledgements
We thank HM Heybroek, E Collin, RA Ennos, and EA Gill for valuable comments on the manuscript, and E González, ML Hollingsworth and A Clarke for technical support. This work was supported by the Dirección General para la Biodiversidad (MMARM, Spain). PF-U. was supported with a FPI scholarship co-funded by the Consejería de Educación (Comunidad de Madrid) and the European Social Fund. MV was supported with a PIF scholarship from the Technical University of Madrid. In memoriam of Margarita Burón Barrio to whom the SECBP owes so much.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article
Cite this article
Fuentes-Utrilla, P., Venturas, M., Hollingsworth, P. et al. Extending glacial refugia for a European tree: genetic markers show that Iberian populations of white elm are native relicts and not introductions. Heredity 112, 105–113 (2014). https://doi.org/10.1038/hdy.2013.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.81
Keywords
This article is cited by
-
Population history, genetic variation, and conservation status of European white elm (Ulmus laevis Pall.) in Poland
Annals of Forest Science (2022)
-
Breeding and scientific advances in the fight against Dutch elm disease: Will they allow the use of elms in forest restoration?
New Forests (2019)
-
Dissection of leaf morphological traits from isolated and declined relict populations of Ulmus laevis reveals putative random ecotype evolution
Plant Systematics and Evolution (2016)
