
Abstract
The comimetic Heliconius butterfly species pair, H. erato and H. melpomene, appear to use a conserved Mendelian switch locus to generate their matching red wing patterns. Here we investigate whether H. cydno and H. pachinus, species closely related to H. melpomene, use this same switch locus to generate their highly divergent red and brown color pattern elements. Using an F2 intercross between H. cydno and H. pachinus, we first map the genomic positions of two novel red/brown wing pattern elements; the G locus, which controls the presence of red vs brown at the base of the ventral wings, and the Br locus, which controls the presence vs absence of a brown oval pattern on the ventral hind wing. The results reveal that the G locus is tightly linked to markers in the genomic interval that controls red wing pattern elements of H. erato and H. melpomene. Br is on the same linkage group but approximately 26 cM away. Next, we analyze fine-scale patterns of genetic differentiation and linkage disequilibrium throughout the G locus candidate interval in H. cydno, H. pachinus and H. melpomene, and find evidence for elevated differentiation between H. cydno and H. pachinus, but no localized signature of association. Overall, these results indicate that the G locus maps to the same interval as the locus controlling red patterning in H. melpomene and H. erato. This, in turn, suggests that the genes controlling red pattern elements may be homologous across Heliconius, supporting the hypothesis that Heliconius butterflies use a limited suite of conserved genetic switch loci to generate both convergent and divergent wing patterns.
Similar content being viewed by others

Introduction
A major goal of modern research in evolutionary biology is to identify the genes and specific nucleotide variation responsible for adaptation (Hoekstra and Coyne, 2007; Stern and Orgogozo, 2008). A related follow-up is then to determine whether the same genes are used repeatedly to generate convergent and divergent phenotypes among closely related taxa (Gompel and Carroll, 2003; Hoekstra and Nachman, 2003; Sucena et al., 2003; Wittkopp et al., 2003; Shapiro et al., 2004; Colosimo et al., 2005; Mundy, 2005; Protas et al., 2006; Kingsley et al., 2009; Steiner et al., 2009). The bold wing patterns of Heliconius butterflies offer a powerful system to answer these questions because there has been incredible phenotypic diversification within the group, combined with convergence among species because of natural selection for Müllerian mimicry (Brown, 1981; Joron et al., 2006 a). However, much of this diversity is controlled by a small number of large effect, Mendelian switch loci (Sheppard et al., 1985; Gilbert, 2003; Jiggins et al., 2005; Kapan et al., 2006). Recent work has positionally cloned genomic regions responsible for a number of these switch loci and shown that across the genus, loci with similar effects on wing pattern are located in similar positions on homologous chromosomes (Kronforst et al., 2006 a; Joron et al., 2006b). For instance, comparative analyses (Baxter et al., 2008) have shown that the same narrow genomic interval of a few hundred kilobases houses the switch loci that control red wing pattern elements in distantly related co-mimics, H. erato (HeD locus) and H. melpomene (HmB and HmD loci). Furthermore, association and expression data point to the same gene, Kinesin, as a potential candidate in both species (Baxter et al., 2010; Counterman et al., 2010).
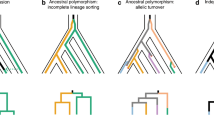
Across Heliconius species and subspecies, the shape, size and position of red wing pattern elements are highly variable as is the hue, which can be brown, orange, crimson or pink (Figure 1a). Although convergent red phenotypes appear to have a conserved genetic basis in Heliconius, it remains unknown whether phenotypically divergent forms use the same genetic machinery to generate entirely novel red pattern elements. Previously, we showed that the presence of red vs brown at the ventral base of the wings in H. cydno and H. pachinus, a Mendelian trait called the G locus (Figure 1b), maps to the same chromosome as the B/D loci of H. erato and H. melpomene (Kronforst et al., 2006a). Furthermore, in crosses between H. melpomene and H. cydno, part of the G phenotype (the thin red line at the base of the fore wing) showed no evidence of recombination with the large red band on the H. melpomene fore wing, HmB, indicating that these traits are very tightly linked or controlled by the same gene (Naisbit et al., 2003). Here we use genetic mapping and fine-scale analyses of genetic differentiation and linkage disequilibrium (LD) to show that the G locus of H. cydno and H. pachinus may be homologous to the B/D loci of H. erato and H. melpomene.

The genomic position of loci controlling red/brown color pattern elements is conserved across Heliconius butterflies. (a) Photos of vental wing surfaces show red and brown pattern elements. The G locus controls the presence of red vs brown (codominant) at the base of the ventral wings, Br controls the presence (dominant) vs absence (recessive) of the brown oval pattern on the ventral hind wing and B controls the presence (dominant) vs absence (recessive) of the red band on the fore wing of H. melpomene. (b) Segregation at linked loci G and Br in an F2 intercross between H. cydno and H. pachinus. (c) Mapping G and Br loci, relative to conserved markers, revealed that the G locus is tightly linked to the same candidate interval housing the B/D loci of H. melpomene and the D locus of H. erato. Unlabeled horizontal lines on linkage maps indicate positions of other linked markers. Comparative data for H. melpomene and H. erato are from Baxter et al. (2008, 2010).
Materials and methods
Genetic mapping
We genotyped an F1 and F2 intercross between H. cydno galanthus and H. pachinus at 44 Amplified fragment length polymorphism markers, one microsatellite and three gene-based markers on the chromosome containing the G locus. Two of the gene-based markers were single-nucleotide polymorphisms, in genes contained within the B/D candidate interval, Ashwin (gene 3 in Figure 2) and Sine Oculis (gene 10 in Figure 2). We also scored each F2 individual for two wing pattern traits, G and Br. The G locus is a Mendelian trait, characterized by the presence of brown vs red pigmentation at the base of the ventral fore- and hind wing (Figure 1b). Heterozygotes are variable but show both colors. The Br locus is a second Mendelian trait that is characterized by the presence vs absence of the brown oval pattern seen on the ventral hind wing of H. cydno. The anterior side of the Br oval is often obscured by the action of the unlinked Yb locus so the trait was scored based on the presence vs absence of the posterior portion. Heterozygotes show the brown oval pattern. We determined the order of markers on the G linkage group and the positions of G and Br relative to these using Joinmap 3.0 (Van Ooijen and Voorrips, 2001) and Mapmaker/Exp 3.0 (Lincoln et al., 1993) using methods described previously (Kronforst et al., 2006 a, 2006 c).

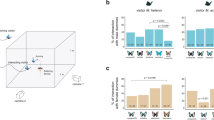
Surveys of genetic differentiation and linkage disequilibrium (LD) through the G locus candidate interval in comparisons among H. cydno, H. pachinus and H. melpomene. For each of 116 polymorphisms (Supplementary Table S3), we calculated FST values in comparisons between (a) H. cydno and H. pachinus, (b) H. cydno and H. melpomene, and (c) H. pachinus and H. melpomene. Red points indicate markers with significant (P<0.05) FST values. Marker order: 1, DNAJ/HSP; 2, Popeye; 3, Ashwin; 4, Sin-Ex; 5, Slu7; 6, Kinesin; 7, GPCR; 8, Esterase; 9, Epoxide hydrolase; 10, Sine Oculis; 11, LRR; 12, Strabismus/Van Gogh; 13, SCY-1; 14, THAP; 15, Helicase (Supplementary Table S1). LD heat maps for (d) H. cydno, (e) H. pachinus and (f) H. cydno and H pachinus together, with similar plots for H. melpomene and pairwise comparisons with H. melpomene in Supplementary Figure S1. White rows/columns indicate comparisons involving monomorphic positions or missing data.
Population genetic analyses
We PCR amplified and sequenced a total of 18 genomic segments from 15 genes spanning the B/D interval as well as a portion of MRSP, a linked gene that lies outside of the G candidate interval, and Caspase, an unlinked control gene (Supplementary Table S1). For each region, we generated comparative DNA sequence data for 16 H. cydno galanthus, 16 H. pachinus and 16 H. melpomene rosina individuals from Costa Rica (Supplementary Table S2). We assembled sequence data into contigs using Lasergene SeqMan 8 (DNASTAR, Madison, WI, USA) and identified and scored SNPs and polymorphic indels by eye. We calculated polymorphism-specific FST values in all pairwise comparisons of the three species using Arlequin 3.0 (Excoffier et al., 2005). To test for evidence of enhanced genetic differentiation within the candidate region, relative to the rest of the genome, we compared average marker FST values for G-linked markers, including MRSP, to 15 unlinked control genes sequenced previously (Kronforst et al., 2006b, 2007) plus Caspase (Supplementary Table S1) using Mann–Whitney U-tests. This test was chosen because FST values were not normally distributed in some comparisons. Our final data sets consisted of approximately 12 kb of comparative DNA sequence data for G-linked markers and 9 kb of DNA sequence data for control genes. Finally, we estimated composite LD (r2) among all polymorphic sites within and between species using the method of Weir (1996) as implemented in Counterman et al. (2010).
Results and discussion
To examine the potential homology among HeD, HmB/D and H. cydno/pachinus G, we first mapped G in a cydno × pachinus F2 brood, relative to markers in the B/D interval (Figures 1b and c). Markers in the interval and G were tightly linked (0 recombinants in 65 individuals). In addition, we also mapped a second wing pattern trait, the Br locus, which controls the brown oval pattern on the ventral hind wing of H. cydno. This switch locus was known to be syntenic with the B/D/G loci and approximately 20 cM away (Naisbit et al., 2003). That placement was confirmed by 17 recombinants between G and Br out of 65 individuals (Figure 1c).
To examine fine-scale patterns of association through the B/D/G candidate interval, we next performed a population genetic screen, comparing SNP and indel allele frequencies among H. cydno, H. pachinus and H. melpomene (Supplementary Table S2). H. cydno and H. pachinus diverged approximately 0.5 Myr to match different mimicry models, and their common ancestor split from H. melpomene approximately 1–1.5 Myr (Kronforst et al., 2006b). Despite displaying highly divergent wing patterns, these closely related and interfertile species hybridize and experience interspecific gene flow across Costa Rica (Kronforst et al., 2006 b; Kronforst, 2008). Compared with data for unlinked control genes, there was evidence for elevated differentiation between H. cydno and H. pachinus within the interval (mean FST, interval genes=0.222, control genes=0.126, U=217.5, P=0.032). Comparisons with H. melpomene did not reveal significantly elevated differentiation in the interval (melpomene vs cydno: mean FST, interval genes=0.452, control genes=0.337, U=179, P=0.126, melpomene vs pachinus: mean FST, interval genes=0.496, control genes=0.337, U=180, P=0.223) likely because of high-background genetic differentiation.
Marker-by-marker analyses of differentiation through the interval revealed no obvious hotspot of divergence in any pairwise comparison of the three species (Figures 2a–c). There were fixed differences (FST=1) throughout the interval in comparisons involving H. melpomene, and moderate differentiation throughout the interval in the comparison between H. cydno and H. pachinus. Although there were no obvious peaks of differentiation in the cydno/pachinus comparison, the polymorphism with the highest FST (0.568) was in epoxide hydrolase (gene 9), which is in the cluster of genes housing the B/D candidate gene, Kinesin (gene 6). Analyses of LD revealed an area of enhanced LD covering the region from genes 5 to 9 (Figures 2d–f), visible most clearly in H. pachinus but also seen in H. cydno. This finding is consistent with a similar pattern of elevated LD in the same area in H. melpomene (Baxter et al., 2010).
Overall, our data did not reveal a defined region of association in the candidate interval, but the general increase in differentiation in the interval between H. cydno and H. pachinus, as well as the signature of enhanced LD around the candidate gene cluster, support the hypothesis that this region has experienced selection and may house the variation responsible for alternate G phenotypes. This lack of a defined signal of differentiation is in sharp contrast to the strong, localized differentiation centered around genes 5–9 in comparisons between geographic subspecies of H. melpomene that differ at the B locus (Baxter et al., 2010). It is also different from the localized signature of differentiation observed between H. cydno and H. pachinus at the position of another mimicry locus, Yb (Baxter et al., 2010). The Yb locus, which controls the presence or absence of a yellow band on the hind wing, is located on a different chromosome and again appears to be homologous across H. melpomene, H. cydno, H. pachinus and H. erato. In the Yb interval, there is a defined hotspot of differentiation between H. cydno and H. pachinus, centered on a leucine-rich repeat gene, but comparisons between subspecies of H. melpomene show generally elevated differentiation in the interval but no isolated hotspot of association. The reasons for these different signatures of selection among species and patterning genes are currently unclear. However, the variation may reflect differences in (1) the strength or timing of selection, (2) the amount of background divergence and LD among hybridizing taxa and/or (3) the complexity of the underlying genetic basis of different traits (that is, whether one or multiple-linked sites contribute to generating the phenotype). Future work, focused on characterizing the functional variation responsible for alternate phenotypes at each of these color-patterning loci, will eventually allow us to tease these potential explanations apart.
A larger question in the field of evolutionary genetics is whether the same genetic mechanisms and pathways are used repeatedly to generate convergent phenotypes. In Heliconius, natural selection for Müllerian mimicry has resulted in radical wing pattern diversification among subspecies and closely related species, coupled with convergence among distantly related species to produce multiple, independent origins of nearly identical wing patterns. The results presented here for the G locus complement and extend other recent work showing that all Heliconius butterflies appear to use the same, limited suite of major switch genes to generate color pattern diversity, both convergent and divergent (Kronforst et al., 2006 a; Joron et al., 2006 b; Baxter et al., 2008, 2010; Papa et al., 2008; Counterman et al., 2010). If this is true, it suggests a variety of important follow-up questions. For instance, over what evolutionary distance is this genetic architecture conserved? In addition, do all Heliconius use the same switch genes because these are the only (or easiest) ways to alter wing pattern or is it a product of their recent common ancestry? Expanding the scope of comparisons with other taxa will be essential to answer these questions and some intriguing hints already exist. For instance, Beldade et al. (2009) recently mapped the genomic positions of multiple lab-generated wing pattern mutants in the butterfly Bicyclus anyana, and found that four of these mutants localized to three linkage groups known to contain Heliconius mimicry switch genes. Although still preliminary, these emerging comparative data suggest that the genomic intervals targeted repeatedly by natural selection in Heliconius may have far-reaching effects on patterning and pigmentation across Lepidoptera.
References
Baxter SW, Nadeau NJ, Maroja LS, Wilkinson P, Counterman BA, Dawson A et al. (2010). Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in the Heliconius melpomene clade. PLoS Genet 6: e1000794.
Baxter SW, Papa R, Chamberlain N, Humphray SJ, Joron M, Morrison C et al. (2008). Convergent evolution in the genetic basis of Müllerian mimicry in Heliconius butterflies. Genetics 180: 1567–1577.
Beldade P, Saenko SV, Pul N, Long AD (2009). A gene-based linkage map for Bicyclus anynana butterflies allows for a comprehensive analysis of synteny with the Lepidopteran reference genome. PLoS Genet 5: e1000366.
Brown KS (1981). The biology of Heliconius and related genera. Annu Rev Entomol 26: 427–456.
Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J et al. (2005). Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307: 1928–1933.
Counterman BA, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP et al. (2010). Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genet 6: e1000796.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform 1: 47–50.
Gilbert LE (2003). Adaptive novelty through introgression in Heliconius wing patterns: evidence for shared genetic ‘tool box’ from synthetic hybrid zones and a theory of diversification. In: Boggs CL, Watt WB, Ehrlich PR (eds). Ecology and Evolution Taking Flight: Butterflies as Model Systems. University of Chicago Press, Chicago, IL. pp 281–318.
Gompel N, Carroll SB (2003). Genetic mechanisms and constraints governing the evolution of correlated traits in drosophilid flies. Nature 424: 931–935.
Hoekstra HE, Coyne JA (2007). The locus of evolution: evo devo and the genetics of adaptation. Evolution 61: 995–1016.
Hoekstra HE, Nachman MW (2003). Different genes underlie adaptive melanism in different populations of rock pocket mice. Mol Ecol 12: 1185–1194.
Jiggins CD, Mavarez J, Beltran M, McMillan WO, Johnston JS, Bermingham E (2005). A genetic linkage map of the mimetic butterfly Heliconius melpomene. Genetics 171: 557–570.
Joron M, Jiggins CD, Papanicolaou A, McMillan WO (2006a). Heliconius wing patterns: an evo-devo model for understanding phenotypic diversity. Heredity 97: 157–167.
Joron M, Papa R, Beltran M, Chamberlain N, Mavarez J, Baxter S et al. (2006b). A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol 4: e303.
Kapan DD, Flanagan NS, Tobler A, Papa R, Reed RD, Gonzalez JA et al. (2006). Localization of Müllerian mimicry genes on a dense linkage map of Heliconius erato. Genetics 173: 735–757.
Kingsley EP, Manceau M, Wiley CD, Hoekstra HE (2009). Melanism in Peromyscus is caused by independent mutations in Agouti. PLoS One 4: e6435.
Kronforst MR (2008). Gene flow persists millions of years after speciation in Heliconius butterflies. BMC Evol Biol 8: 98.
Kronforst MR, Kapan DD, Gilbert LE (2006a). Parallel genetic architecture of parallel adaptive radiations in mimetic Heliconius butterflies. Genetics 174: 535–539.
Kronforst MR, Salazar C, Linares M, Gilbert LE (2007). No genomic mosaicism in a putative hybrid butterfly species. Proc Biol Sci 274: 1255–1264.
Kronforst MR, Young LG, Blume LM, Gilbert LE (2006b). Multilocus analyses of admixture and introgression among hybridizing Heliconius butterflies. Evolution 60: 1254–1268.
Kronforst MR, Young LG, Kapan DD, McNeely C, O’Neill RJ, Gilbert LE (2006c). Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc Natl Acad Sci USA 103: 6575–6580.
Lincoln SE, Daly MJ, Lander ES (1993). MAPMAKER/EXP. Whitehead Institute for Biomedical Research, Boston.
Mundy NI (2005). A window on the genetics of evolution: MC1R and plumage colouration in birds. Proc Biol Sci 272: 1633–1640.
Naisbit RE, Jiggins CD, Mallet J (2003). Mimicry: developmental genes that contribute to speciation. Evol Dev 5: 269–280.
Papa R, Martin A, Reed RD (2008). Genomic hotspots of adaptation in butterfly wing pattern evolution. Curr Opin Genet Dev 18: 559–564.
Protas ME, Hersey C, Kochanek D, Zhou Y, Wilkens H, Jeffery WR et al. (2006). Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nat Genet 38: 107–111.
Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jonsson B et al. (2004). Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 428: 717–723.
Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC (1985). Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Phil Trans R Soc Lond B 308: 433–610.
Steiner CC, Rompler H, Boettger LM, Schoneberg T, Hoekstra HE (2009). The genetic basis of phenotypic convergence in beach mice: similar pigment patterns but different genes. Mol Biol Evol 26: 35–45.
Stern DL, Orgogozo V (2008). The loci of evolution: how predictable is genetic evolution? Evolution 62: 2155–2177.
Sucena E, Delon I, Jones I, Payre F, Stern DL (2003). Regulatory evolution of shavenbaby/ovo underlies multiple cases of morphological parallelism. Nature 424: 935–938.
Van Ooijen JW, Voorrips RE (2001). JoinMap 3.0, Software for the Calculation of Genetic Linkage Maps. Plant Research International, Wageningen, The Netherlands.
Weir BS (1996). Genetic Data Analysis II: Methods for Discrete Population Genetic Data. Sinauer, Sunderland, MA.
Wittkopp PJ, Williams BL, Selegue JE, Carroll SB (2003). Drosophila pigmentation evolution: divergent genotypes underlying convergent phenotypes. Proc Natl Acad Sci USA 100: 1808–1813.
Acknowledgements
We thank Brian Counterman for assistance calculating LD. This project was funded by the NIH NIGMS grant GM068763 to MRK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Chamberlain, N., Hill, R., Baxter, S. et al. Comparative population genetics of a mimicry locus among hybridizing Heliconius butterfly species. Heredity 107, 200–204 (2011). https://doi.org/10.1038/hdy.2011.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2011.3

{kind=link}