
Abstract
Gene transfer of potent immunostimulatory cytokines such as interleukin-12 (IL-12) is a potential treatment for advanced cancer. Different vectors and IL-12 modifications have been developed to avoid side effects associated with high serum levels of the cytokine, while preserving its antitumor properties. Here we have evaluated two alternative strategies using the Syrian hamster as a model for pancreatic cancer metastatic to the liver. Local administration of an oncolytic adenovirus (OAV) expressing a single-chain version of IL-12 caused transient, very intense elevations of IL-12 in serum, resulting in severe toxicity at sub-therapeutic doses. Anchoring IL-12 to the membrane of infected cells by fusion with the transmembrane domain of CD4 reduced systemic exposure to IL-12 and increased the tolerance to the OAV. However, only a modest increase in the therapeutic range was achieved because antitumor potency was also reduced. In contrast, systemic administration of a helper-dependent adenoviral vector (HDAd) equipped with a Mifepristone-inducible expression system allowed sustained and controlled IL-12 production from the liver. This treatment was well tolerated and inhibited the progression of hepatic metastases. We conclude that HDAds are safer than OAVs for the delivery of IL-12, and are promising vectors for immunogene therapy approaches against pancreatic cancer.
Similar content being viewed by others


Introduction
Different gene therapy (GT) approaches can be applied to stimulate immune responses against cancer cells. Local production of immunostimulatory cytokines in the tumor microenvironment or the surrounding tissues is one of them.1 As the main objective is to improve the biodistribution and pharmacokinetics of the cytokine, the performance of vectors is a key factor for the safety and efficacy of these strategies. In the case of viral vectors, it has been postulated that local inflammation can contribute to trigger an efficient antitumor response.2 This effect can be greatly enhanced if the virus is able to replicate in and destroy cancer cells (oncolytic virus, OV), through a combination of direct tumor destruction, release of tumor antigens, danger signals and strong expression of the transgene.3 Proof of concept for the efficacy of immunovirotherapy is already available in the clinic. In a recent phase III clinical trial, an oncolytic herpes simplex virus armed with granulocyte-macrophage colony-stimulating factor (T-VEC) showed superior durable response rates in advanced melanoma patients compared with subcutaneous administration of the recombinant granulocyte-macrophage colony-stimulating factor protein.4 An oncolytic vaccinia virus expressing the same transgene (Pexa-Vec) has shown promising results in phase II clinical trials for hepatocellular carcinoma.5 In this case, intratumoral administration of the virus led to an increase in serum concentrations of granulocyte-macrophage colony-stimulating factor and elevation of neutrophil counts in peripheral blood. Although no adverse events were directly attributed to this systemic effect, it is clear that intratumoral administration of vectors does not prevent release of transgene products in the circulation. This issue is particularly important if potent immunostimulatory cytokines are incorporated in OVs.6 A good example is interleukin-12 (IL-12). This cytokine is able to stimulate the proliferation and cytolytic activity of natural killer and T cells, apart from its anti-angiogenic effect.7 Most of its biological effects are mediated by stimulation of interferon-γ (IFNγ) production. Robust antitumor effects and the ability to cooperate with adoptive cell therapies are important attributes of IL-12.8 However, systemic exposure needs to be tightly controlled in order to avoid a severe, potentially lethal inflammatory syndrome observed when the recombinant protein was administered intravenously at high doses.9 When IL-12 is incorporated as a therapeutic gene in a GT vector, the two subunits of this heterodimeric cytokine can be expressed from a polycistronic IL-12 mRNA, or as a fusion protein in which both subunits are linked by a short flexible domain (single-chain, scIL-12).10 OVs encoding IL-12 have demonstrated a strong antitumor effect in pre-clinical models of different cancers,6, 11, 12 including a model of experimental liver metastases from pancreatic cancer in Syrian hamsters.13 These animals are especially suited for the evaluation of oncolytic adenoviruses (OAVs) because they are immunocompetent, permissive for human adenovirus replication14, 15 and sensitive to IL-12 side effects.13 Controlling the release of IL-12 from infected cells is challenging, as the intensity of expression depends not only on the efficacy of infection but also on the replication of the virus in tumor cells. This leads to heterogeneity in serum concentrations of IL-12 among individuals,13 and greatly reduces the possibility to perform dose escalation of the virus. Membrane anchoring of cytokines has been proposed as a method to obtain high local concentrations without extensive release into circulation.16 Fusion of p35 and p40 subunits with glycosylphosphatidylinositol (GPI) anchor signals achieves attachment of IL-12 in the membrane of transfected cells.17 In the same line, a scIL-12 construct fused with the transmembrane and cytoplasmic domains of the B7-1 protein is expressed as a type I transmembrane protein in the cells and retains its immunostimulatory functions.18 In fact, when the transgene was incorporated in a replication-deficient adenoviral vector, a potent antitumor effect was observed with negligible elevations of IL-12 in the serum of mice. The ability of these IL-12 modifications to increase the safety of OVs has never been investigated.
An alternative approach to exploit the antitumor properties of IL-12 is the use of long-term expression vectors carrying drug-inducible expression systems. In this case, direct oncolysis and release of tumor antigens are not part of the mechanism of action, but this may be compensated by the possibility of controlling the duration and intensity of IL-12 expression. Doxycycline and Mifepristone-inducible systems have been used to control the expression of IL-12, delivered in vivo by adeno-associated19 and helper-dependent adenoviral vectors (HDAd).20, 21 Both vectors showed excellent safety profile and efficacy in a syngeneic murine model of colorectal cancer liver metastases. The stability of transgene expression allows the design of tailored induction regimes adapted as adjuvants for standard treatments such as chemotherapy.21
In this work, we provide a side by side comparison between new OAVs carrying different membrane-anchored scIL-12 variants and a previously described HDAd vector equipped with a liver-specific Mifepristone-inducible IL-12 expression system (HC-Ad/RUmIL-12).21 Safety and efficacy of both approaches were studied in an aggressive model of pancreatic ductal adenocarcinoma (PDA) metastatic to the liver in Syrian hamsters.14
Results
Modifications in the IL-12 expression cassette to reduce secretion of the cytokine
Transgene expression mediated by OAVs is very intense in pancreatic cancer cells because of the efficiency of transduction and the replication of viral genomes inside the cells.14 Although the design of these agents favors replication in cancer cells and the intratumoral route of administration minimizes infection in off-target tissues, transduction of highly sensitive organs such as liver cannot be completely avoided.13 The risk of liver-derived secretion of IL-12 is especially relevant in the case of local administration of OAVs in hepatic metastases. To decrease this potential source of toxicity, we incorporated the target sequence of a liver-specific microRNA (miR-122)22, 23 into the 3′ UTR of the scIL-12. This construct was designated scIL-12miR (see Figure 1). We chose scIL-12 instead of the native form because this compact complementary DNA is compatible with the size constraints of the OAV genome.13 In addition, modifications in the scIL-12 were incorporated to anchor the cytokine to the membrane of expressing cells. To this end, two different strategies were evaluated. Both of them rely on the carboxy-terminal fusion of scIL-12 with protein domains associated with the cell membrane: GPI anchor signal from the folate receptor (scIL-12-GPI)24 and a transmembrane domain from human CD4 (IL-12-TM).25, 26 These constructs were incorporated in a eukaryotic expression plasmid driven by a cytomegalovirus (CMV) promoter and transfected in target cells from hamster PDA (HaP-T1) and human cells from a well-differentiated hepatocellular carcinoma expressing miR-122 (HuH-7).22 Quantification of IL-12 released in the supernatant of cells revealed that the incorporation of the miR-122 target sequence caused a drastic reduction in IL-12 production in HuH-7 cells, whereas a slight, nonsignificant decrease was observed in HaP-T1 cells (Figure 2a). This correlates with the high content of miR-122 in HuH-7 cells compared with HaP-T1 cells (Supplementary Figure 1a). Regarding scIL-12 modifications, fusion with TM was efficient in preventing IL-12 secretion, in contrast with the GPI anchor signal. When the presence of IL-12 in the membrane of cells was analyzed by flow cytometry (Figures 2b and c), scIL-12 was detected in approximately 50% HaP-T1 cells transfected with the pC-scIL-12-TM plasmid, in good correlation with transfection efficiency in these cells. In fact, when the analysis was performed in cells highly permissive for transfection (HEK-293), the percentage was close to 80% (Figure 2b). Interestingly, scIL-12-GPI was also detected in the surface, suggesting that this method of membrane anchoring results in a dynamic equilibrium between attachment and shedding of the cytokine.27 The binding to glycolipids was demonstrated by the reduction of membrane-bound scIL-12-GPI observed upon treatment with phosphatidylinositol phospholipase C, as shown in Figure 2b.

Schematic representation of GT vectors used in this study. The upper part corresponds to the four different OAVs and the lower part corresponds to the HDAd. ΔE1A, deletion of the CR2 domain of the E1A gene; ΔE3, partial deletion of the E3 region; GAL4-E1Bp, inducible promoter comprising four Gal4-binding sites and a TATA box derived from the adenoviral E1B gene; GPI, GPI anchor signal; HPRT and C346, fragments from the human hypoxanthine-guanine phosphoribosyltransferase and C346 cosmid used as DNA stuffer, respectively; IRES, internal ribosomal entry site; ITR, inverted terminal repeat; ψ, packaging signal; p40 and p35, heavy and light chains of IL-12, respectively; miR, target sequence for the miR-122; pA, polyadenylation signal; TM, transmembrane domain from human CD4; TTRp, transthyretin promoter; GLP65, transactivator composed of a Gal4 DNA-binding domain, a human progesterone receptor ligand binding domain and a human p65 activator domain. Not drawn to scale.

Membrane anchoring of scIL-12 in transfected cells. HuH-7, HaP-T1 or HEK-293 cells were transfected with the indicated plasmids. A subset of cells transfected with pC-scIL-12-GPI was treated with phosphatidylinositol phospholipase C (PI-PLC), as indicated. (a) Twenty-four hours later, conditioned media were collected for quantification of scIL-12 by ELISA. Cells were then harvested, stained with an antibody against murine IL-12 and analyzed by flow cytometry. (b) Percentage of cells displaying scIL-12 in their surface. (c) Representative flow cytometry histograms corresponding to HaP-T1 cells. *P<0.05, **P<0.01, ***P<0.001.
scIL-12-TM retains biological activity
Fusion of scIL-12 with the CD4 transmembrane domain caused a drastic modification in its secretion pattern. To verify that scIL-12-TM retained its immunostimulatory properties, we analyzed its ability to stimulate the production of IFNγ on lymphocytes. To this end, HEK-293 cells were transfected with pC-scIL-12miR or pC-scIL-12-TM, and incubated with mouse splenocytes for 1 h. Splenocytes were then separated from HEK-293 cells, washed and maintained for additional 48 h until mRNA was extracted to determine the expression of IFNγ by quantitative reverse transcriptase-PCR (qRT-PCR). We observed that cells expressing both versions of scIL-12 were equally efficient in stimulating IFNγ (Figure 3a). In addition, when splenocytes were incubated with fixed amounts of scIL-12 or scIL-12-TM produced from transfected cells, expression of IFNγ was equivalent in both cases. In fact, a nonsignificant increase in potency was observed in the case of scIL-12-TM (Figure 3b). The activation of murine and Syrian hamster splenocytes was similar (Figure 3c), confirming the biological effect of murine IL-12 on both species.

scIL-12-TM retains biological activity. HEK-293 cells were transfected with pC-scIL-12-miR, pC-scIL-12-TM or empty plasmids (control). (a) Twenty-four hours later, transfected cells were incubated with murine splenocytes for 1 h. Splenocytes were then separated from HEK-293 cells, washed and maintained for additional 48 h until mRNA was extracted to analyze expression of IFNγ by qRT-PCR. (b) scIL-12 concentration was determined in conditioned media from transfected cells, and the indicated amounts (horizontal axis) were used to stimulate murine splenocytes. Expression of murine IFNγ was analyzed by qRT-PCR 48 h later. (c) Mouse or hamster splenocytes were stimulated with supernatants containing 10 ng ml–1 of scIL-12. In all cases, values correspond to the relative IFNγ mRNA content, using β-actin as an internal reference and applying the formula 2(Ct βactin-IFNγ)x10 000. **P<0.01.
Evaluation of IL-12 modifications in the context of OAVs
Once we had analyzed the function of the modified scIL-12 constructs, we incorporated them into the genome of a standard OAV (Figure 1). In this viral backbone, a widely used deletion in the CR2 domain of the E1A early gene confers preferential replication in cancer cells.28, 29 A deletion in the E3 region (6.7 K/gp19 K genes) was used to accommodate the transgenes. In this genome location, expression is controlled by endogenous viral promoters and is associated with viral replication, as previously described.30 When HaP-T1 cells were infected at relatively low multiplicities of infection (MOI) with viruses carrying the different scIL-12 modifications, cytokine secretion and membrane attachment recapitulated our findings in transfected cells (Figures 4a and b). This means that viral infection does not alter the properties the anchored cytokines, so we could find scIL-12-GPI both in the supernatant and the membrane of infected cells, whereas scIL-12-TM is predominantly displayed in the cell surface. However, when we increased the MOI of the OAV-scIL-12-TM virus, we observed that the capacity of membrane anchoring can be saturated at a MOI of 20 infectious units (i.u.) per cell (Figure 4c). Under these circumstances, secretion of scIL-12 is similar between OAV-scIL-12-miR and OAV-scIL-12-TM. This is not due to oncolysis, as no decrease in cell viability was observed between infected and control cells at the moment of analysis (24 h, data not shown). This result suggests that the performance of anchored cytokines in the context of replication-competent vectors may be different from non-viral or defective viral vectors, and careful in vivo testing is required.

Membrane anchoring of scIL-12 in cells infected with OAVs. HaP-T1 cells were infected with the indicated OAVs at MOI 2. Supernatants and cells were collected separately 24 h later. (a) Concentration of scIL-12 in conditioned media. (b) Percentage of cells displaying scIL-12 in their surface, determined by flow cytometry. (c) HaP-T1 cells were infected with OAV-scIL-12-miR (squares) or OAV-scIL-12-TM (inverted triangles) at the indicated MOIs, and the concentration of IL-12 in the conditioned media was determined 24 h later. **P<0.01, ***P<0.001.
Function of the miR-122 target site in vivo
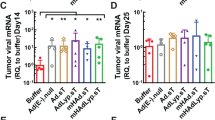
Previous works have demonstrated that the Syrian hamster is a suitable model to evaluate the efficacy and toxicity of OAVs,14, 15 especially in the context of immunovirotherapy approaches.13 In this case, we used an aggressive pancreatic cancer liver metastases model based on the intrahepatic implantation of HaP-T1 cells.14 These carcinogen-induced PDA cells harbor K-ras mutations and give rise to rapidly progressing tumors resembling many clinical and histological characteristics of human neoplasms.31 Untreated animals require to be killed 5–6 weeks after cell implantation because of high tumor burden. Replication and oncolytic effect of human adenovirus in HaP-T1 cells is similar than the observed in human PDA cell lines (Supplementary Figure 2). Analysis of liver and tumor samples from Syrian hamsters confirmed preferential expression of miR-122 in normal liver (Supplementary Figure 1b), although the magnitude of the difference (100-fold) was lower than that observed between HuH-7 and HaP-T1 cells in vitro. The first parameter evaluated in vivo was the ability of the miR-122 target site to inhibit the expression of scIL-12 in the liver of Syrian hamsters. To this end, OAV-scIL-12 and OAV-scIL-12-miR were administered intravenously or intratumorally and the animals were killed 24 h later to determine the concentration of the cytokine in serum, as well as liver and tumor extracts. As shown in Figure 5a, hamsters treated intravenously with OAV-scIL-12-miR had a 98% reduction in IL-12 concentration in serum, in good correlation with the reduction in liver extracts. This trend was also observed in tumor extracts, but in this case the quantification of IL-12 may be highly influenced by the amount present in blood. In fact, when the viruses were administered locally, no differences in intratumoral IL-12 content were observed between OAV-scIL-12 and OAV-scIL-12-miR (Figure 5b). The relative reduction of IL-12 concentration in serum and liver of hamsters that receive intratumoral OAV-scIL-12-miR may reflect the activity of viruses that reach circulation from the tumor and then infect the liver. To better determine vector biodistribution, viral genomes were quantified in liver and tumor samples of hamsters treated by OAV-scIL-12 (Supplementary Figure 3). As expected, intratumoral administration of the virus reduces infection of normal liver, but it does not prevent it completely. Of note, efficient infection of tumors was obtained upon intravenous administration. Analysis of other organs by qRT-PCR revealed that the transgene was expressed almost exclusively in liver and tumors (data not shown). In summary, the miR-122 target site was able to significantly reduce the expression of scIL-12 from the liver.

Incorporation of the target sequence for miR-122 in the 3′ UTR of scIL-12 reduces expression of the cytokine in the liver. Syrian hamsters bearing intrahepatic HaP-T1 tumors were injected intravenously (a) or intratumorally (b) with 109 i.u. of OAV-scIL-12 or OAV-scIL-12-miR. Twenty-four hours later, the animals were killed for collection of liver, tumor and blood. scIL-12 concentration was quantified by ELISA in serum (expressed as ng ml–1, left vertical axis) and in tissue extracts from normal liver or tumors (expressed as ng g–1 tissue, right vertical axis). *P<0.05, **P<0.01.
Toxicity and antitumor effect of OAVs expressing modified IL-12 in vivo
To determine if the scIL-12 modifications increased the therapeutic range of OAVs, we administered different doses of the viruses intratumorally and studied systemic exposure to IL-12, tolerance to the treatment and antitumor effect. In Figure 6a we represent the concentration of scIL-12 in the serum of animals 24 h after the injection of each virus at the indicated doses, which represents the peak concentration in most cases. In good correlation with the in vitro data, the only virus that showed a robust inhibition of scIL-12 release was OAV-scIL-12-TM. Incorporation of the miR-122 target site achieved only a moderate reduction compared with OAV-scIL-12, suggesting that most of the scIL-12 secreted into the bloodstream derives from tumors. As expected, OAV-scIL-12-GPI behaved similar to OAV-scIL-12-miR and was unable to retain the cytokine in the membrane of transduced cells. Nevertheless, this virus was included in the efficacy studies to determine if the combination of anchored and free scIL-12 could be beneficial for the antitumor effect. In all cases, a dose-dependent increase in scIL-12 concentration was observed. This had a clear correlation with toxicity (Figure 6b). All viruses were lethal at 109 i.u. per hamster (60–80% mortality), except for OAV-scIL-12-TM. Even in this case, this dose cannot be considered completely safe because the next dose escalation step (2 × 109 i.u.) caused a 30% mortality rate. As described,13 the most common cause of death in hamsters expressing scIL-12 was lung edema (Figure 6c). Of note, viral replication does not seem to have a predominant role in toxicity because a replication-competent adenovirus with wild type E1A gene expressing the reporter gene luciferase (Ad-WT-Luc) was well tolerated at the maximal dose tested (2 × 109 i.u.). The analysis of scIL-12 kinetics (Figure 6d) indicates that increasing the intratumoral dose of OAV-scIL-12-TM not only raises the peak concentration of the cytokine, but also prolongs its presence in serum. From 2.5 × 108 to 1 × 109 i.u., the maximum concentration was observed 24 h after infection and then it rapidly decreases. The same occurs with the rest of viruses tested (data not shown). In contrast, at higher doses of OAV-scIL-12-TM (2 × 109 i.u. and beyond), scIL-12 concentration is maintained at least for 3 days. This is compatible with a transient amplification of the virus in tumors and may contribute to toxicity. When the antitumor effect of all viruses was evaluated at a safe dose (2.5 × 108 i.u.), only a small inhibition of tumor progression was observed 1 month after initiation of treatment (Figure 7a). The only group that showed a significant reduction in tumor volume without mortality was OAV-scIL-12-TM at 109 i.u. (Figure 7b). However, dose escalation was not possible, as shown in Figure 6b. When the OAV-scIL-12-TM virus was administered intravenously, mortality reached 50% at 109 i.u. per hamster (data not shown), and no antitumor effect was observed at 2.5 × 108 i.u. per hamster (Figure 7c). Therefore, membrane anchoring of scIL-12 using the CD4 TM domain achieves a modest increase in the therapeutic index of OAVs when they are administered locally.

Intratumoral administration of OAV-scIL-12-TM reduces systemic exposure to scIL-12 and is less toxic than OAVs carrying other scIL-12 variants. Syrian hamsters bearing intrahepatic HaP-T1 tumors were treated with a single local administration of the indicated OAVs at different doses. (a) Blood was collected 24 h later to determine scIL-12 concentration. The dose of each virus is plotted in the horizontal axis of the graph. (b) Survival of animals 3 weeks after initiation of treatment. At this point, only treatment-related deaths were observed. White squares represent hamsters treated with 2 × 109 i.u. Ad-WT-Luc. (c) Representative microphotograph of paraffin-embedded lung samples from a hamster treated with 1 × 109 i.u. OAV-scIL-12. Hematoxylin–eosin staining (x200). Note the intense inflammatory infiltration and the accumulation of fluid in alveoli of the treated animal (asterisks), characteristic of lung edema. (d) Serum concentration of scIL-12 over time in hamsters treated with OAV-scIL-12-TM at the following doses: 2.5 × 108 i.u. (dotted gray line), 1 × 109 i.u. (solid gray line), 2 × 109 i.u. (dotted black line) and 5 × 109 (solid black line).

Antitumor effect of OAVs expressing scIL-12 variants. Syrian hamsters bearing intrahepatic HaP-T1 tumors were treated with a single local administration of the indicated OAVs at 2.5 × 108 i.u. (a) or 1 × 109 i.u. per hamster (b). The same model was used to evaluate the effect of OAV-scIL-12-TM upon intravenous administration (c) at the indicated doses. In all cases, hamsters were killed 4 weeks after the initiation of treatment and tumor volumes were measured at necropsy. Individual tumor volumes and group medians are represented. Note that values from animals that did not survive at this point as a result of scIL-12 toxicity were not included in this graph. *P<0.05.
Controlled expression of IL-12 in the liver mediated by an HDAd is safe and reduces the progression of hepatic metastases
The limitations observed with membrane anchoring of scIL-12 in the context of OAVs prompted us to evaluate an alternative approach to obtain safe gene transfer of IL-12 in our PDA model. The HC-Ad/RUmIL-12 vector is designed for liver-specific expression of IL-12 in response to the inducer Mifepristone (RU486).20 In contrast with OAVs, the high-cloning capacity of this vector allows the accommodation of the inducible system and the simultaneous expression of the p35 and p40 IL-12 subunits as independent polypeptides. No major differences have been observed between the biological effect of the natural heterodimeric IL-12 and the fusion protein scIL-12 in vitro.10 However, the formation of p40 homodimers could have a modulatory effect. To clarify this point we performed a direct in vivo comparison between IL-12 and scIL-12. First-generation adenoviral vectors expressing each cytokine version under the control of a constitutive CMV promoter were administered intravenously to C57BL/6 mice. Concentrations of IL-12 and its main effector IFNγ were analyzed in the serum of animals (Supplementary Figure 4). No major differences in the ability to stimulate IFNγ expression were observed among both IL-12 constructs, suggesting that their biological activity is equivalent. In agreement with this finding, toxicity of Ad-CMV-IL-12 and Ad-CMV-scIL-12 was similar (50% mortality at 1 × 108 i.u. per mouse, data not shown). The therapeutic effect of HC-Ad/RUmIL-12 was evaluated in hamsters following the protocol depicted in Figure 8a. Once PDA tumors were established in the liver, animals received an intravenous injection of clodronate liposomes to deplete Kupffer cells and facilitate hepatocyte transduction with adenovirus.32 This treatment does not affect the progression of tumors (data not shown). Twenty-four hours later, the vector was administered intravenously at 109 or 5 × 108 i.u. per hamster. A third group remained untreated. Seven days after infection, IL-12 expression was stimulated by daily intraperitoneal administration of Mifepristone. The dose of the inducer was adjusted to maintain therapeutic levels of the cytokine, as previously described.21 In the case of hamsters, serum concentration of IL-12 above 5000 ng ml–1 for >3 days causes weight loss and should be avoided to prevent severe toxicity (data not shown). The minimum concentration of IL-12 required for a therapeutic effect will depend on the characteristics of the tumor and the animal species. When HC-Ad/RUmIL-12 was administered at 109 i.u. per hamster, initial induction with 250 μg kg–1 Mifepristone caused a strong elevation of IL-12 in serum (exceeding 5000 ng ml–1 in some hamsters), but levels could be modulated by Mifepristone adjustment in subsequent days (Figure 8b) and all animals tolerated the 9-day induction protocol. One month after the first induction, hamsters were killed and tumor volumes were determined at necropsy. A significant antitumor effect was observed (Figure 8c), with complete tumor eradication in 2/6 cases and inhibition of tumor progression in another two hamsters. Although the target organ for IL-12 toxicity in hamsters is the lung, we analyzed standard biochemical parameters of liver damage because this organ is exposed to the highest concentrations of the cytokine in this experimental setting. As shown in Figures 8d and e a peak of transaminases and bilirubin was observed during the first week, consistent with the elevated concentration of IL-12 produced in the initial phase of the induction regime. This toxic effect was reversible and responded to the reduction of Mifepristone doses. Nevertheless, the second treatment group (5 × 108 i.u. HC-Ad/RUmIL-12) was intended to study if lower IL-12 concentrations maintained for longer periods of time were equally effective. Initial induction revealed a 100-fold reduction in IL-12 expression compared with the high-dose group (Figure 8b), compatible with the non-linear dose-response effect characteristic of adenoviral transduction in the liver. In this case, intensification of the induction regime was needed to keep detectable IL-12 for 18 days, albeit at low levels (Figure 8b). Importantly, this protocol achieved an antitumor effect equivalent to the high-dose group (Figure 8c), with 4/8 disease-free hamsters at the end of the experiment. No alterations in biochemical parameters attributable to the treatment were observed (Figures 8d and e). Note that the presence of tumor in the liver of hamsters causes progressive elevation of transaminases in the absence of treatment, reaching 258±134 U l–1 5 weeks after cell implantation. These results indicate that sustained IL-12 expression in the liver is a safe option to control progression of PDA liver metastases.

Antitumor effect and modulation of IL-12 production in hamsters treated with HC-Ad/RUmIL-12. (a) Schematic representation of the experiment. A single dose of HC-Ad/RUmIL-12 vector was administered intravenously (i.v.) 5 days after intrahepatic (i.h.) inoculation of HaP-T1 cells. Treatment groups included high dose (1 × 109 i.u. per hamster) and low dose (5 × 108 i.u. per hamster) of vector. A third group was left untreated (saline injection). One week later, induction of IL-12 expression was performed by daily intraperitoneal (i.p.) administrations of Mifepristone (Mif). Initiation of induction is referred as ‘day 0’ of the experiment. The doses of Mifepristone in the high-dose group were (in μg kg–1): 250 for days 1–2; 125 for days 3–6 and 250 for days 7–9. In the low-dose group the doses were: 250 for days 1–2; 1000 for days 3–10; 4000 for days 11–14 and 8000 for days 15–18. (b) Serum concentration of scIL-12 at the indicated times (high vector dose in black line and low vector dose in gray line). (c) Tumor volumes 4 weeks after initiation of the induction protocol. Individual values and group medians are represented. (d) Concentration of transaminase (alanine aminotransferase) in serum at the indicated days. (e) Concentration of bilirubin in serum. *P<0.05, ***P<0.001.
Discussion
The vector of choice for an immunogene therapy approach depends on the characteristics of the therapeutic gene (potency, toxicity) and the mechanism of action being pursued. IL-12 is a multifunctional cytokine that can have important roles in different strategies, from classical gene transfer to virotherapy11, 12, 13 or adoptive cell therapies.8 The Syrian hamster model used in this work offers the opportunity to compare, under the same experimental conditions, the efficacy and safety of IL-12 expressed from OAVs and HDAds. In the case of OAVs, the potential synergy between the oncolytic effect and the action of IL-12 requires relatively high viral doses administered locally in the tumor. The objective is to obtain a vaccination effect against tumor antigens with a limited number of vector administrations. Dose fractionation is not a good option, as abundant laboratory and clinical evidences suggest that the rapid onset of neutralizing antibodies is a major barrier for adenovirus redosing.33, 34 Therefore, the challenge is to restrict the release of IL-12 from the tumors. Here we provide evidence that a standard scIL-12 is not an adequate transgene because meaningful antitumor effects are always associated with severe side effects in this aggressive PDA model. A single dose of 5 × 108 i.u. OAV-scIL-12 caused a 20% mortality rate. This amount of virus is far below the minimum needed to obtain a relevant oncolytic effect with OAVs lacking therapeutic genes. In our experience, doses above 109 i.u. of replication-competent adenoviruses expressing reporter genes were largely inefficient in PDA hamster models based on H2T13 and HaP-T1 cells (data not shown). Others have described antitumor effects in a subcutaneous renal cancer model in hamsters only when they administer 5 doses of 1010 i.u. OAV spanning 1 week, before the onset of neutralizing antibodies.35 The characteristics of our advanced PDA model pose an additional challenge because the hepatic location of tumors facilitates infection of hepatocytes and off-target expression of IL-12 that would be readily secreted into the bloodstream. Our viral biodistribution analysis confirmed that intratumoral administration of the virus reduces infection of normal liver. This correlates with a decrease in hepatic IL-12 content, which is further inhibited by the incorporation of the miR-122 target sequence into the 3′UTR of the expression cassette in the OAV-scIL-12-miR virus. However, only a partial reduction in serum scIL-12 and toxicity were achieved. This reinforces the concept that secretion of scIL-12 from tumors is largely responsible for the toxicity. Interestingly, we found that intravenous administration of the virus is as efficient as the local injection in terms of tumor infection. This is in contrast with previous observations in extrahepatic tumors,33 and suggests that pancreatic cancer metastases in the liver can be targeted by systemic administration of OAVs. However, the elevated infection of normal liver could compromise the safety of this route of administration. Membrane anchoring of IL-12 and other cytokines has shown the ability to restrict the release of these proteins from cells transfected ex vivo17, 36 or when defective viral vectors are used in vivo.18 We have confirmed that fusion of scIL-12 with the transmembrane domain of CD4 is an efficient way to anchor the cytokine in the membrane of cells and to reduce its secretion, while maintaining the biological effect. In fact, this was the only modification in the scIL-12 cassette that reduced significantly the toxicity of the OAV. To the best of our knowledge, this is the first report of a membrane-anchored cytokine in the context of an OV. We found advantages and limitations of this strategy. The OAV-scIL-12-TM virus achieved a significant antitumor effect without severe adverse effects, and this is a clear improvement versus other agents, including OAV-scIL-12-GPI. However, the increase in therapeutic range was limited by two circumstances. In one hand, the anchoring of the cytokine is saturated when expression is very intense in the context of viral replication, and the cytolytic effect can contribute to the release of scIL-12 still attached to cell membranes. On the other hand, the cell-bound scIL-12 was not sufficient to protect animals from tumor progression in the absence of detectable levels of the cytokine in serum in our animal model. If cancer cell-bound scIL-12 had a marked increase in its potency, we would expect an improvement in the therapeutic index of OAV-scIL-12-GPI virus, which is not the case. Of note, the OAV-scIL-12-TM virus becomes toxic at 2 × 109 i.u. per hamster, when serum scIL-12 levels are still lower than those causing toxicity with the other OAVs. However, we cannot conclude that this form of scIL-12 is more toxic, because a concomitant extension in the duration of transgene expression occurred at high viral doses. This is compatible with amplification of the virus in the tumors, but is still far from the dose of OAV needed to exert a potent oncolytic effect. In summary, our data using membrane-anchored IL-12 provide evidence that this approach can improve the properties of an armed OV, but the case of OAVs is especially challenging. Adenovirus combines a very efficient transduction efficacy with relatively low direct oncolytic potency. We believe other OVs with the opposite profile such as HSV-1 or vaccinia could greatly benefit from membrane-anchored cytokine expression.
In clear contrast with OAVs, the management of HC-Ad/RUmIL-12 was easier. To aid in the interpretation of our results, we have performed an in vivo side by side comparison of murine IL-12 and scIL-12 in their natural host. Our data indicate an equivalent biological potency and toxicity of both cytokine versions. This is in agreement with the biological activity originally described in vitro,10 in contrast with other reports proposing a stronger effect of scIL-12 based on indirect evidences.37 The efficient dose-dependent control of IL-12 levels by Mifepristone alleviates the need to define a precise dose of vector with the optimum balance between safety and efficacy. This level of control compensates for the lack of linearity in hepatic transduction observed when different doses of the vector were administered systemically. Surprisingly, this threshold effect occurred despite pre-treatment with clodronate liposomes, suggesting that other barriers apart from Kupffer cells prevent a free access of adenovirus to hepatocytes in hamsters. We are currently investigating if direct intrahepatic injection of the virus overcomes this hurdle. Apart from the increase in hepatocyte transduction, clodronate liposomes can reduce acute toxicity of adenoviral vectors by dampening inflammatory responses.38 Therefore, we believe that clinically compatible ways to avoid the interaction of adenoviral particles with macrophages should be considered in clinical trials to increase the safety of these vectors. Once the target concentration of IL-12 is obtained, it has been described that maintaining it for long periods of time in mice requires adjustment of the inducer dose. This is in part due to the IFNγ-dependent silencing of the liver-specific promoter used in the inducible system.21 In the case of hamsters, long-term expression from HC-Ad/RUmIL-12 can also be compromised by immune responses against the murine transgene present in this vector. Despite this technical limitation, we provide evidence that a significant therapeutic effect can be obtained with low level IL-12 expression for 18 days, and this protocol is at least as efficient as high cytokine concentrations for short periods. In summary, we conclude that immunogene therapy approaches based on IL-12 may be safe and efficient against hepatic metastases of PDA if the transgene is delivered to the liver by an HDAd vector and the expression of the cytokine is controlled by a drug-inducible system.
Materials and methods
Plasmids and transfection
The murine scIL-12 gene10 was obtained by PCR from the pSFG-mIL-12 plasmid (kindly provided by Dr. Mulligan, Harvard Medical School, Boston, MA, USA) and introduced into the pCDNA3 plasmid for further modifications (pC-scIL-12). The target site for miR-122 was obtained by hybridization of the following oligonucleotides: 5′-CTAGACAAACACCATTGTCACACTCCAT-3′ and 5′-CTAGATGGAGTGTGACAATGGTGTTTGT-3′, and the cassette was introduced in the 3′UTR of scIL-12 to give rise to the plasmid pC-scIL-12-miR. The GPI anchor signal from the human folate receptor (MSGAGPWAAWPFLLSLALMLLWLLS)24 was linked to the carboxy-terminal end of scIL-12 using a flexible peptide from the pyruvate ferredoxin oxidoreductase (APAETKAEPMT) as a bridge. To this end, the following overlapping oligonucleotides: 5′-GTACTGGCGCGCCGGCACCAGCAGAAACAAAAGCAGAACCAATGACAATGAGTGGGGCTGGGCCCTGGGCAGCCTGGCC-3′ and 5′-GTACTGCGGCCGCTCAGCTGAGCAGCCACAGCAGCATTAGGGCCAGGCTAAGCAGGAAAGGCCAGGCTGCCCAGGG-3′ were annealed, filled with DeepVent Polymerase and introduce in the pC-scIL-12miR plasmid to generate the pC-scIL-12-GPI plasmid. The TM domain from human CD4 (MALIVLGGVAGLLLFIGLGIFFCV)25 was fused to scIL-12 using the same linker (plasmid pC-scIL-12-TM). To this end, a cassette based on the following oligonucleotides: 5′-GTACTGGCGCGCCGATGGCCCTGATTGTGCTGGGTGGCGTCGCCGGCCTCCTGCTTTTCATTGGGCTAGGCATCTTCTTCTGTGTCtgaGCGGCCGCgtact-3′ and 5′-AGTACGCGGCCGCTCAGACACAGAAGAAGATGCCTAGCCCAATGAAAAGCAGGAGGCCGGCGACGCCACCCAGCACAATCAGGGCCATCGGCGCGCCAGTAC-3′ was used as a template for PCR amplification with an external pair of oligonucleotides: 5′-GTACTGGCGCGCCGGCACCAGCAGAAACAAAAGCAGAACCAATGACAATGGCCCTGATTGTGCTG-3′ and 5′-AGTACGCGGCCGCTCAGACACAGAAG-3′. Transfections were carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), following the instructions provided by the manufacturer. The cassettes containing scIL-12 were subcloned in a shuttle plasmid (pGL3Cla-Nar) and then introduced into a plasmid containing the genome of an OAV with a unique ClaI site into the E3 region (pSDCla). All molecular biology enzymes were from New England Biolabs (Ipswich, MA, USA) unless otherwise stated.
Viruses
The viral sequence present in pSDCla is based on human adenovirus type 5, which was cloned using PacI sites in a plasmid containing the kanamycin resistance gene. Preferential replication in cancer cells is obtained by partial deletion of the E1A gene (comprising bp 922 to 947), as previously described.28, 29 Transgenes were incorporated into the E3 region using the unique ClaI site inserted in place of the of the E3-6.7 K/gp19K genes (deletion comprising bp 28 555 to 29 355 in the viral genome). This location determines that expression is under the control of endogenous viral promoters and is dependent on viral replication.30 Plasmids were digested with PacI to release the viral genomes, and were transfected in HEK-293 cells for virus rescue. The Ad-WTLuc virus39 is based on wild-type adenovirus type 5 (Ad5) with insertion of the luciferase gene into the same E3 region. The Ad-WT-CMV-GFP virus contains an expression cassette comprising the CMV promoter and the EGFP gene inserted into the E3 region of wild-type adenovirus. In this case, transgene expression does not require virus replication. Ad-CMV-IL-12 and Ad-CMV-scIL-12 are replication-deficient (first-generation, E1/E3-deleted) adenoviral vectors carrying an expression cassette for IL-12 and scIL-12, respectively, under the control of the CMV promoter. All viruses were amplified in HEK-293 cells and purified by ultracentrifugation in CsCl gradients. Quantification of i.u. was done using the Adeno-X rapid titer kit (Clontech, Mountain View, CA, USA). HC-Ad/RUmIL-12 is an HDAd (also called high-capacity or ‘gutless’) vector carrying a Mifepristone-inducible system for the expression of p35 and p40 subunits of murine IL-12. Both IL-12 subunits are co-expressed because of an internal ribosomal entry site. The transactivator of this system is expressed under the control of a liver-specific promoter (transthyretin). Production and amplification of this vector has been previously described.40 The ratio i.u. per viral particles of OAV-scIL-12, OAV-scIL-12-miR, OAV-scIL-12-GPI, OAV-scIL-12-TM, Ad-WT-Luc and HC-Ad/RUmIL-12 batches used in this study were 136, 136, 94, 129, 81 and 39, respectively.
Cells
The HEK-293 cells (ATTC CRL-1573; ATCC, Manassas, VA, USA) and the tumor cell lines HuH-7 (human hepatocellular carcinoma, JCRB Genebank, Osaka, Japan), HeLa (human cervical cancer, ATCC#CCL-2), PANC-1 (human PDA ATCC#CRL-1469) and HaP-T1 (Syrian hamster PDA, German Collection of Microorganisms and Cell Cultures DSMZ ACC 222) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum supplemented with 100 U ml–1 penicillin, 100 μg ml–1 streptomycin and 2 mM L-glutamine. Reagents are from Gibco (Gaithersburg, MD, USA). All cells were maintained at 37 °C with 5% CO2 in a humidified incubator. All cells were kept mycoplasma free.
Quantification of miR-122 expression
A stem-loop reverse transcription approach was used to quantify miR-122 expression in HaP-T1 cells and tissues, as previously described.41 HeLa and HuH-7 cells were used as negative and positive controls, respectively.42 Total RNA was extracted from cells using TRI Reagent (Sigma, St Louis, MO, USA). A RT-PCR step was performed using the primer 5′-GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACCAAACA-3′. The product was used as a template for a qPCR using a miR-122-specific forward primer 5′-TTCCTTGGAGTGTGACAATGG-3′ and a universal reverse primer 5′-GTGCAGGGTCCGAGGT-3′. Standard curve for miR-122 was obtained using the oligonucleotide 5′-GTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACCAAACACCATTGTCACACTCCA-3′. In the case of tissue extracts, cell content was normalized by comparison with hamster β-actin mRNA determined by qRT-PCR using primers 5′-GTCGTACCACTGGCATTGTG-3′ (forward) and 5′-GTCACGCACAATTTCCCTCT-3′ (reverse).
In vitro viability assays
Cells were seeded in 24-well tissue culture plates at 2 × 104 cells per well and 24 h later they were infected with serial dilution of viruses. Cell viability was determined 5 days after infection by crystal violet staining. The percentage of survival is calculated by comparison with uninfected cells. Data were analyzed using the GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
Flow cytometry
Cells were harvested and fixed with 0.25% paraformaldehyde for 5 min. Staining was performed with rat anti-Mouse IL-12 (p40/p70) and then with a fluorescein isothiocyanate-labeled secondary antibody (mouse anti-rat IgG1), both from BD Biosciences (San Diego, CA, USA). All antibodies were incubated 10 min at 4 °C. Appropriate isotype controls were used to verify staining specificity. Cells were acquired on a FACSCalibur and analyzed using FlowJo (Tree Star Inc., Ashland, OR, USA).
Cytokine quantification
Murine IFNγ concentration in serum and murine IL-12 concentrations in cell supernatant, serum or liver lysates were determined by OptE1A mouse IL-12 (p70) and murine IFNγ ELISA kits, respectively, both from BD Bioscience. qRT-PCR for analysis of IL-12 expression in liver extracts was performed with the following set of primers: 5′-ATGATGACCCTGTGCCTTGG-3′ (forward) and 5′-ACTCTGTAAGGGTCTGCTTCTC-3′ (reverse). qRT-PCR for IFNγ in splenocytes was performed with primers 5′-GGCCATCCAGAGGAGCATAG-3′ (forward) and 5′-CCATGCTGCTGTTGAAGAAGTTAG-3′ (reverse) in hamsters and primers 5′-GCATATCTGGAGGAACTGGC-3′ (forward) and 5′-CGACTCCTTTTCCGCTTCCT-3′ (reverse) in mice.
In vitro viral replication assay (determination of virus burst size)
Cells (5 × 104 per well) were cultured in 24-well plates overnight and then were infected in Dulbecco’s modified Eagle’s medium containing 2% fetal bovine serum with Ad-WT-CMV-Luc at MOIs 50 and 100 for HaP-T1 and PANC-1, respectively, to achieve an infection rate close to 100%. The virus was removed 4 h later, cells were washed twice with phosphate-buffered saline and incubated in Dulbecco’s modified Eagle’s medium supplemented with 2% fetal bovine serum for 48 h. Then cells were lysed by three cycles of freezing and thawing and viruses were quantified by the Adeno-X Rapid titer kit (BD Bioscience).
Tumor model and in vivo treatments
Liver metastases of pancreatic cancer were established in Syrian (Golden) hamsters (Mesocricetus Auratus; HSD HAN: AURA, 5 weeks of age from Harlan, Indianapolis, IN, USA) by intrahepatic injection of 3 × 106 HaP-T1 cells through laparotomy, as previously described.14 When tumors reached at least 200 mm3 (typically 2 weeks after cell implantation), local administration of OAVs were performed by direct injection through laparotomy. Average pre-treatment tumor volume was equivalent in all experimental groups. Intravenous inoculation of HDAd was performed by retro-orbital injection. One day before HDAd administration, hamsters received an intravenous injection of 500 μl clodronate liposomes (Clodlip B.V., Amsterdam, The Netherlands) to obtain transient depletion of Kupffer cells. Mifepristone (RU486, Sigma) was administered intraperitoneally dissolved in sesame oil at daily doses of 125–8000 μg kg–1, as previously described.21 Tumor volumes were calculated at necropsy using the formula V= (Dxd2)/2, where D and d are the major and minor diameters, respectively. Necropsies were performed by an investigator without information about group identity of animals. Ad-CMV-IL-12 and Ad-CMV-scIL-12 were administered intravenously in C57BL/6 mice. Serum concentration of IL-12 and IFNγ were determined during the first week after injection, and survival was followed for at least 1 month. No animals were excluded from the analysis. All procedures were carried out following protocols approved by the local ethical committee in accordance with recommendations for proper care and use of laboratory animals.
Quantification of genome copies in tissues
Liver and tumor samples were frozen in liquid nitrogen and DNA was isolated using QIAmp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Two μl of each sample were used for quantification of viral copies. PCR was performed using iCycler iQ Real-Time Detection System (Hercules, CA, USA) with SYBR green fluorophore. The primer sequences used for amplification of Adenovirus E4 gene were 5′-CTAACCAGCGTAGCCCCGA-3′ (forward) and 5′-TGAGCAGCACCTTGCATTTT-3′ (reverse). Reactions were performed in a total volume of 20 μl including 10 μl 2X iQ SYBR Green Supermix (Bio-Rad, Singapore), 0.6 μl of each primer at the concentration of 10 μM and 2 μl of template. Parameters of PCR included an initial denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 15 s, annealing at 60 °C for 30 s, extension at 72 °C for 30 s and one cycle of 72 °C for 4 min. Melt analysis was performed to determinate the specificity of the PCR amplification. To calculate the amount of copies per mg tissue, a standard curve of purified Ad5 genomes was prepared.
Statistical analysis
The Mann–Whitney test was applied for statistical comparison of two groups. Comparisons of more than two groups were performed by analysis of variance with Bonferroni correction, using the GraphPad Prism program (GraphPad Software). Unless otherwise stated, triplicate samples were used for in vitro experiments. Error bars in graphs correspond to s.e.m. For animal studies, sample size estimate was obtained from http://www.biomath.info/power/index.htm. The number of animals per group is indicated in the scatter plot graphs. Unless otherwise stated, all experiments were repeated at least twice.
References
Jinushi M, Tahara H . Cytokine gene-mediated immunotherapy: current status and future perspectives. Cancer Sci 2009; 100: 1389–1396.
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC . Going viral with cancer immunotherapy. Nat Rev Cancer 2014; 14: 559–567.
Tong AW, Senzer N, Cerullo V, Templeton NS, Hemminki A, Nemunaitis J . Oncolytic viruses for induction of anti-tumor immunity. Curr Pharm Biotechnol 2012; 13: 1750–1760.
Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, Spitler LE, Puzanov I, Doleman S, Ye Y, Vanderwalde AM, Coffin R, Kaufman H . OPTiM: a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol 2013; 31 (Suppl): aLBA9008.
Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013; 19: 329–336.
Varghese S, Rabkin SD, Liu R, Nielsen PG, Ipe T, Martuza RL . Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther 2006; 13: 253–265.
Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res 2007; 13: 4677–4685.
Kerkar SP, Leonardi AJ, van Panhuys N, Zhang L, Yu Z, Crompton JG et al. Collapse of the tumor stroma is triggered by IL-12 induction of Fas. Mol Ther 2013; 21: 1369–1377.
Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997; 90: 2541–2548.
Lieschke GJ, Rao PK, Gately MK, Mulligan RC . Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat Biotechnol 1997; 15: 35–40.
Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci USA 2013; 110: 12006–12011.
Jarnagin WR, Zager JS, Klimstra D, Delman KA, Malhotra S, Ebright M et al. Neoadjuvant treatment of hepatic malignancy: an oncolytic herpes simplex virus expressing IL-12 effectively treats the parent tumor and protects against recurrence-after resection. Cancer Gene Ther 2003; 10: 215–223.
Bortolanza S, Bunuales M, Otano I, Gonzalez-Aseguinolaza G, Ortiz-de-Solorzano C, Perez D et al. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in syrian hamsters. Mol Ther 2009; 17: 614–622.
Bortolanza S, Alzuguren P, Bunuales M, Qian C, Prieto J, Hernandez-Alcoceba R . Human adenovirus replicates in immunocompetent models of pancreatic cancer in Syrian hamsters. Hum Gene Ther 2007; 18: 681–690.
Thomas MA, Spencer JF, La Regina MC, Dhar D, Tollefson AE, Toth K et al. Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res 2006; 66: 1270–1276.
Cheng TL, Roffler S . Membrane-tethered proteins for basic research, imaging, and therapy. Med Res Rev 2008; 28: 885–928.
Nagarajan S, Selvaraj P . Glycolipid-anchored IL-12 expressed on tumor cell surface induces antitumor immune response. Cancer Res 2002; 62: 2869–2874.
Pan WY, Lo CH, Chen CC, Wu PY, Roffler SR, Shyue SK et al. Cancer immunotherapy using a membrane-bound interleukin-12 with B7-1 transmembrane and cytoplasmic domains. Mol Ther 2012; 20: 927–937.
Vanrell L, Di Scala M, Blanco L, Otano I, Gil-Farina I, Baldim V et al. Development of a liver-specific Tet-on inducible system for AAV vectors and its application in the treatment of liver cancer. Mol Ther 2011; 19: 1245–1253.
Wang L, Hernandez-Alcoceba R, Shankar V, Zabala M, Kochanek S, Sangro B et al. Prolonged and inducible transgene expression in the liver using gutless adenovirus: a potential therapy for liver cancer. Gastroenterology 2004; 126: 278–289.
Gonzalez-Aparicio M, Alzuguren P, Mauleon I, Medina-Echeverz J, Hervas-Stubbs S, Mancheno U et al. Oxaliplatin in combination with liver-specific expression of interleukin 12 reduces the immunosuppressive microenvironment of tumours and eradicates metastatic colorectal cancer in mice. Gut 2011; 60: 341–349.
Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA 2007; 104: 12884–12889.
Suzuki T, Sakurai F, Nakamura S, Kouyama E, Kawabata K, Kondoh M et al. miR-122a-regulated expression of a suicide gene prevents hepatotoxicity without altering antitumor effects in suicide gene therapy. Mol Ther 2008; 16: 1719–1726.
Paladino S, Lebreton S, Tivodar S, Campana V, Tempre R, Zurzolo C . Different GPI-attachment signals affect the oligomerisation of GPI-anchored proteins and their apical sorting. J Cell Sci 2008; 121: 4001–4007.
Li Z, Blissard GW . Functional analysis of the transmembrane (TM) domain of the Autographa californica multicapsid nucleopolyhedrovirus GP64 protein: substitution of heterologous TM domains. J Virol 2008; 82: 3329–3341.
Wu L, Zhou T, Yang ZY, Svehla K, O'Dell S, Louder MK et al. Enhanced exposure of the CD4-binding site to neutralizing antibodies by structural design of a membrane-anchored human immunodeficiency virus type 1 gp120 domain. J Virol 2009; 83: 5077–5086.
Poloso NJ, Nagarajan S, Mejia-Oneta JM, Selvaraj P . GPI-anchoring of GM-CSF results in active membrane-bound and partially shed cytokine. Mol Immunol 2002; 38: 803–816.
Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000; 19: 2–12.
Johnson L, Shen A, Boyle L, Kunich J, Pandey K, Lemmon M et al. Selectively replicating adenoviruses targeting deregulated E2F activity are potent, systemic antitumor agents. Cancer Cell 2002; 1: 325–337.
Hawkins LK, Johnson L, Bauzon M, Nye JA, Castro D, Kitzes GA et al. Gene delivery from the E3 region of replicating human adenovirus: evaluation of the 6.7K/gp19 K region. Gene Therapy 2001; 8: 1123–1131.
Abraham AT, Shah SR, Davidson BR . The HaP-T1 Syrian golden hamster pancreatic cancer model: cell implantation is better than tissue implantation. Pancreas 2004; 29: 320–323.
Schiedner G, Hertel S, Johnston M, Dries V, van Rooijen N, Kochanek S . Selective depletion or blockade of Kupffer cells leads to enhanced and prolonged hepatic transgene expression using high-capacity adenoviral vectors. Mol Ther 2003; 7: 35–43.
Bunuales M, Garcia-Aragoncillo E, Casado R, Quetglas JI, Hervas-Stubbs S, Bortolanza S et al. Evaluation of monocytes as carriers for armed oncolytic adenoviruses in murine and Syrian hamster models of cancer. Hum Gene Ther 2012; 23: 1258–1268.
Liu TC, Hwang T, Park BH, Bell J, Kirn DH . The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther 2008; 16: 1637–1642.
Thomas MA, Spencer JF, Toth K, Sagartz JE, Phillips NJ, Wold WS . Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther 2008; 16: 1665–1673.
Ji J, Li J, Holmes LM, Burgin KE, Yu X, Wagner TE et al. Synergistic anti-tumor effect of glycosylphosphatidylinositol-anchored IL-2 and IL-12. J Gene Med 2004; 6: 777–785.
Waehler R, Ittrich H, Mueller L, Krupski G, Ameis D, Schnieders F . Low-dose adenoviral immunotherapy of rat hepatocellular carcinoma using single-chain interleukin-12. Hum Gene Ther 2005; 16: 307–317.
Alzuguren P, Hervas-Stubbs S, Gonzalez-Aseguinolaza G, Poutou J, Fortes P, Mancheno U et al. Transient depletion of specific immune cell populations to improve adenovirus-mediated transgene expression in the liver. Liver Int 2015; 35: 1274–1289.
Bortolanza S, Bunuales M, Alzuguren P, Lamas O, Aldabe R, Prieto J et al. Deletion of the E3-6.7K/gp19K region reduces the persistence of wild-type adenovirus in a permissive tumor model in Syrian hamsters. Cancer Gene Ther 2009; 16: 703–712.
Gonzalez-Aparicio M, Mauleon I, Alzuguren P, Bunuales M, Gonzalez-Aseguinolaza G, San Martín C et al. Self-inactivating helper virus for the production of high-capacity adenoviral vectors. Gene Ther 2011; 18: 1025–1033.
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005; 33: e179.
Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol 2004; 1: 106–113.
Acknowledgements
We thank Richard C Mulligan (Department of Genetic, Harvard Medical School, Boston, MA, USA) for the pSFG-mIL-12 plasmid and Stefan Kochanek for the pRS25 plasmid. This work has been funded by grants SAF2009-11324, SAF2012-39578 from the Spanish Department of Science, and the UTE project CIMA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Rights and permissions
About this article

Cite this article
Poutou, J., Bunuales, M., Gonzalez-Aparicio, M. et al. Safety and antitumor effect of oncolytic and helper-dependent adenoviruses expressing interleukin-12 variants in a hamster pancreatic cancer model. Gene Ther 22, 696–706 (2015). https://doi.org/10.1038/gt.2015.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2015.45
This article is cited by
-
Vector engineering, strategies and targets in cancer gene therapy
Cancer Gene Therapy (2022)
-
Optimizing oncolytic virotherapy in cancer treatment
Nature Reviews Drug Discovery (2019)
-
Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent
Nature Communications (2017)
-
Combined therapy with oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly suppressed human hepatocellular carcinoma both in vitro and in an orthotopic transplanted mouse model
Journal of Experimental & Clinical Cancer Research (2016)
-
Oncolytic viruses—immunotherapeutics on the rise
Journal of Molecular Medicine (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}