
Abstract
Protein-losing enteropathy (PLE) is a clinical disorder of protein loss from the gastrointestinal system that results in hypoproteinemia and malnutrition. This condition is associated with a wide range of gastrointestinal disorders. Recently, a unique syndrome of congenital PLE associated with biallelic mutations in the DGAT1 gene has been reported in a single family. We hypothesize that mutations in this gene are responsible for undiagnosed cases of PLE in infancy. Here we investigated three children in two families presenting with severe diarrhea, hypoalbuminemia and PLE, using clinical studies, homozygosity mapping, and exome sequencing. In one family, homozygosity mapping using SNP arrays revealed the DGAT1 gene as the best candidate gene for the proband. Sequencing of all the exons including flanking regions and promoter regions of the gene identified a novel homozygous missense variant, p.(Leu295Pro), in the highly conserved membrane-bound O-acyl transferase (MBOAT) domain of the DGAT1 protein. Expression studies verified reduced amounts of DGAT1 in patient fibroblasts. In a second family, exome sequencing identified a previously reported splice site mutation in intron 8. These cases of DGAT1 deficiency extend the molecular and phenotypic spectrum of PLE, suggesting a re-evaluation of the use of DGAT1 inhibitors for metabolic disorders including obesity and diabetes.
Similar content being viewed by others


Introduction
Protein-losing enteropathy (PLE) has been classified as either an erosive gastrointestinal (GI) disorder, a nonerosive GI disorder, or a disorder involving increased central venous pressure or mesenteric lymphatic obstruction.1 The consequences of PLE include fat malabsorption and fat-soluble vitamin deficiencies. Although the clinical presentation of PLE varies according to the etiology, most patients present with edema and hypoalbuminemia; in PLE, albumin loss into the intestine may reach 60% of the total albumin pool2 (normal, ~15%). PLE falls within the broad spectrum of the congenital diarrheal disorders (CDDs), classified based upon their aberrations in absorption and transport of nutrients and electrolytes, enterocyte differentiation and polarization, enteroendocrine cell differentiation, and modulation of the intestinal immune response.3
Patients with biallelic mutations in DGAT1 (MIM #: 604900) manifest aspects of both PLE and CDD.4 DGAT1 encodes a diacyl CoA:diacylglycerol acyl transferase (DGAT) that converts diacylglycerides to triglycerides (TGs) by adding an acyl CoA moiety.5 The enzyme belongs to the family of membrane-bound O-acyl transferases (MBOATs), with its active domain located in the lumenal region of the endoplasmic reticulum (ER). In the small intestine, DGAT helps to absorb TGs, whereas in the liver it produces TGs from fatty acids synthesized de novo or taken up from the circulation.6
DGAT1 may be a target for antiobesity drugs, based upon studies of a DGAT1-knockout mouse exhibiting resistance to diet-induced obesity, increased energy expenditure, and insulin and leptin sensitivities.7, 8 However, only a single family with DGAT1 mutations has been described. Here we present two additional families with biallelic defects in DGAT1, expanding current knowledge of the spectrum of the disease phenotype and its mutation profile as well as its potential prevalence.
Materials and methods
Patients
Patients were evaluated at the Edmond and Lily Safra Children's Hospital, Sheba Medical Center. Written informed consent was obtained, and the Institutional Review Board approved the genetic studies.
Homozygosity mapping and sequencing
SNP genotyping was performed on genomic DNA from the proband and all members of Family 1, except 1-IV-4 (Figure 1a), using the Human Omniexpress DNA Analysis BeadChip (Illumina, San Diego, CA, USA) and the GenomeStudio software (Illumina). Homozygous areas >75 Mb and segregating only with the proband were examined using the online software, Genomic Oligoarray and a SNP array evaluation tool; candidate genes were selected using an autosomal recessive model filter.9

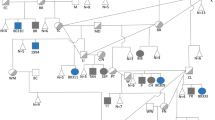
Pedigree of families 1 (a) and 2 (b). Affected patients are depicted in solid/shaded shape. Arrow points to the proband in each family.
For Sanger dideoxy sequencing, primers were designed to cover all coding exons and flanking intronic regions of DGAT1 (NM_012079.5; primer sequences available upon request). Direct sequencing of PCR amplification products was performed using BigDye 3.1 Terminator Chemistry (Applied Biosystems, Woolston, Warrington, UK); products were separated on an ABI 3130xl genetic analyzer (Applied Biosystems, Foster City, CA, USA). Data were evaluated using the Sequencher v5.0 software (Gene Codes Corporation, Ann Arbor, MI, USA). The novel variant identified in this study has been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and has the accession number RCV000201459.
Exome sequencing
One microgram of dsDNA was sheared by sonication (Covaris M220 Instrument, Woburn, MA, USA) to an average size of 200 bp. Library construction was performed on a Wafergen Apollo324 (Fremont, CA, USA) that size-selects fragments by double-SPRI binding with different concentrations of PEG for a high cut and a low cut. After nine cycles of PCR amplification using the Clonetech Advantage II kit (Clontech Laboratories, Inc., Mountain View, CA, USA, 350 ng of genomic library was recovered. Three libraries with different barcodes were pooled before exome enrichment (3-plex) using the NimbleGen EZ Exome V2 kit (Roche, Madison, WI, USA). Library pools were enriched according to the manufacturer’s recommendations and sequenced on an Illumina HiSeq2500, generating ~90 million paired end reads, 125 bases long and each equivalent to 7.5 GB of usable high-quality sequence per sample. We used BWE mem algorithm, as previously described,10 for alignment of the human genome (hg19). GATK version 2.4.7 (Broad Institute, Cambridge, MA, USA) with the UnifiedGenotyper algorithm was applied for variant calling including all steps mentioned in the best practice pipeline.11 KGG-seq12 was used for annotation of detected variants, but in-house scripts were applied for filtering based on family pedigree and intersections.
Cell culture
Fibroblasts were cultured from forearm skin biopsies and grown in high-glucose (4.5 g/l) DMEM supplemented with 15% FBS, 2 mM L-glutamine, nonessential amino-acid solution, and penicillin–streptomycin. Normal adult human dermal fibroblasts (ATCC PCS-201-012) served as controls.
Expression studies
Total RNA was isolated from fibroblasts with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and treated with a DNase kit (DNA-free) according to the manufacturer’s protocol (Life Technologies, Carlsbad, CA, USA). RNA concentration and purity were assessed on a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). First-strand cDNA was synthesized using a high-capacity RNA-to-cDNA kit (Applied Biosystems). Quantitative real-time PCR (qPCR) was performed using Power SYBR Green PCR master mix (Applied Biosystems, Woolston, Warrington, UK) and Bio-Rad qPCR machine (CFX96 Touch Real Time PCR detection system, Philadelphia, PA, USA) with standard qPCR parameters to analyze the expression of DGAT1 compared with the control gene GAPDH (primer sequences available upon request). Results were analyzed with the comparative CT method.13, 14
Western blotting
Fibroblasts were grown for 48 h at 1 × 106 cells per flask in DMEM+10% fetal calf serum. Cell lysates, prepared using lysis buffer (50 mM Tris, pH 7; 150 mM NaCl; 0.1% SDS; 0.5% sodium deoxycholate; 1% Triton x-100; and 1 mM EDTA) supplemented with protease inhibitors (complete, Mini, EDTA-free, Roche), were normalized by the BCA protein assay (Bio-Rad) and subjected to western blotting. Cystosolic fractions were prepared following a rapid, efficient and practical method.15 Samples were electrophoresed on a 12% Tris-glycine gel and blotted onto a 0.2-μm nitrocellulose membrane (Invitrogen, Carlsbad, CA, USA). Membranes were blocked with Li-Cor blocking buffer (Li-Cor Biosciences, Lincoln, NE, USA) overnight at 4 °C, and then incubated with the following antibodies: goat anti-DGAT1 antibody (Sigma, St Louis, MO, USA), mouse-anti-DGAT1 antibody (Abcam, Cambridge, MA, USA), and anti-β-actin (Sigma). After several washes in PBS with 0.1% Tween, membranes were incubated with the appropriate secondary antibodies (Li-Cor Biosciences). Proteins were detected using the Li-Cor imaging system (Li-Cor Biosciences).
Results
Case reports
Family 1
The proband (Patient 1-IV-5, Figure 1a), an 11-month-old male born to consanguineous parents of Arab-Muslim descent, presented with chronic diarrhea, hypoalbuminemia, hypogammaglobulinemia, and failure to thrive (FTT). An older male sibling (Patient 1-IV-4) died at 16 months of age with a similar clinical course consisting of diarrhea, hypoalbuminemia, severe edema, weight loss, and skin abscesses beginning at 4 months of age (Figure 1a). Three older siblings were unaffected.
Patient 1-IV-5 was born at term following an uneventful pregnancy and vaginal delivery; birth weight was 3200 g. The neonate was breast-fed until 2 months, when the infant formula was introduced. Recurrent episodes of watery diarrhea (up to five times a day) occurred despite changing to a soy-based formula. Repeated hospitalizations were required for dehydration due to diarrhea and lower-extremity edema due to hypoalbuminemia (~1.5 g/dl). Serum IgG was 41 mg/dl (normal, 670–1530), IgA was 33 mg/dl (52–274), IgM 44 mg/dl (48–179), and IgE 13.1 IU/ml (4–163). Hemoglobin was 6.3 g/dl (normal, 11.0–18.0), MCV 67 fl (85–123), and serum iron 3.3 mcg/dl (22–148). Sweat chloride, thyroid function tests, serum lipid profile, blood and urine cultures, fecal reducing substances, and T-cell repertoire were normal. An amino-acid-based formula, along with albumin infusions and 10 days of metronidazole, did not improve the symptoms, and intravenous immune globulin was administered monthly for 4 months.
Upon admission to Sheba Medical Center, the infant was cachectic with FTT (weight 5900 g, height 64 cm, head circumference 41 cm, all <3%. The abdomen was mildly distended, and there was digital clubbing and developmental delay; at 11 months he rolled over but could not crawl. The diarrhea did not recur with regular infant formula, but he did require Monogen formula with supplementation of total parenteral nutrition (TPN) and intravenous lipids to gain weight properly. Fasting serum lipids were normal, apart from TGs of 168 mg/dl (normal, 30–150). Serum vitamins A, B1, B6, B12, C, D, and E were normal. Upper GI endoscopy was normal, and scintigraphy ruled out intestinal lymphangiectasia. Fecal alpha-1-antitrypsin was17.0 mg/g dry weight (normal, 0–2.6), but fecal elastase was normal and PLE of unknown etiology was diagnosed. The boy was discharged home free of diarrhea, gaining weight, and with a serum albumin of 4.1 g/dl.
Family 2
Patients 2-III-12 and 2-III-13 (Figure 1b) were among eight full-term children born to a non-consanguineous Ashkenazi-Jewish couple. The two siblings presented with PLE that improved by age 2 years and 6 months, respectively. Two similarly affected cousins (2-III-1 and 2-III-5) were unavailable for evaluation (Figure 1b).
Patient 2-III-12 had a birth weight of 3.97 kg and was breast-fed with supplementary cow’s milk formula. Eight days after birth, he had non-bloody, watery diarrhea, and was diagnosed with an enterobacter urinary tract infection and rotavirus gastroenteritis. At 2 months, he was re-admitted because of FTT, peripheral edema, severe hypoalbuminemia, and hypotonia. Diarrhea continued on an amino-acid-based formula and improved on TPN. At 4 months, he was re-admitted with similar symptoms; repeated stool cultures, rotavirus antigen, and PCR for bacterial pathogens, enterovirus, and adenovirus were negative. The stool was negative for leukocytes. Serum alpha-1-antitrypsin, sweat test, stool elastase, blood amino acids, and urine organic acids were normal; urine-reducing substances were negative. Quantitative immunoglobulins, lymphocyte subpopulations, and responses to mitogens were normal. Stool alpha-1-antitrypsin varied from 10 to 13.5 mg/g (normal, <2.6 mg/g), and the serum albumin was persistently low, leading to a diagnosis of PLE. Duodenal and colonic biopsies showed no evidence of chronic granulomatous disease, autoimmune enteropathy, food protein-induced enterocolitis, microvillous inclusion disease, or tufting enteropathy. Neuroendocrine cells were present in intestinal biopsies. Congenital lymphangiectasia, a cause of PLE, was excluded by endoscopy and histology. TGs and cholesterol levels were unremarkable at 4 months of age, and levels of TG, cholesterol, HDL, LDL, and VLDL remained normal at 6 months of age.
A hospital-designed chicken soup formula mixed with rice water and oral rehydration solution improved the diarrhea and normalized serum albumin. Solid foods were introduced, and the boy was weaned off TPN. He is currently 4.5 years old, with normal growth and development, on a regular diet except for dairy products, which cause diarrhea.
The second affected male sibling (Patient 2-III-13) was born after a normal gestation; birth weight was 4.16 kg. Immediately after birth, a right-sided incarcerated inguinal hernia was noticed; surgery revealed a Meckel’s diverticulum in the incarcerated bowel. After surgery he received TPN, but feeding with breast milk, cow’s milk formula, or an amino-based formula all resulted in watery diarrhea up to eight times per day. Metabolic screen, serum immunoglobulin levels, and head ultrasound were normal, whereas an abdominal ultrasound revealed crossed fused renal ectopia with moderate to marked dilatation of the collecting systems and left hydronephrosis. Stool cultures for bacterial pathogens, rotavirus, enterovirus, and adenovirus were negative. The infant exhibited PLE, with stool alpha-1-antitrypsin levels of 10 mg/g (normal, <2.6 mg/g) and hypoalbuminemia. Duodenal and colon biopsies were negative for autoimmune enteropathy, food protein-induced enterocolitis, microvillous inclusion disease, and tufting enteropathy. Neuroendocrine cells were present in intestinal biopsies. Serum TGs were 260 mg/dl (normal, 30–150) and VLDL was 52 mg/dl (normal, 4–40) with normal levels of LDL, HDL, and total cholesterol at 2 months of age; at 6 months, TG, cholesterol, HDL, LDL, VLDL, apolipoprotein A1, and apolipoprotein B levels were also normal. Fat-soluble vitamins (A, E, and D) were normal at 2 and 4 months. TPN was necessary for weight maintenance.
On oral rehydration therapy plus a chicken soup protein formula mixed with rice water, the diarrhea improved and the serum albumin normalized. After recurrent episodes of methicillin-resistant Staphylococcus aureus (MRSA) infection and sepsis, his central line was removed; he currently maintains his weight, albumin, and hydration on enteral feeds.
SNP analysis and Sanger sequencing in Family 1
SNP arrays revealed 10 areas of homozygosity (Table 1); DGATI, associated with congenital diarrhea and PLE,4 resided in one such area. Sanger sequencing identified a novel homozygous missense variant in exon 10 of DGAT1, NM_012079.4: c.884T>C (Figure 2a), which is predicted to lead to p.(Leu295Pro) in the protein level. Parents and unaffected siblings were carriers, and one unaffected brother was homozygous normal. The p.(Leu295Pro) variant was predicted to be pathogenic (SIFT, score: 0.06; Polyphen-2, score: 1) and was not present in any of the available databases (1000 Genomes, NIH Undiagnosed Diseases Program, Exome Aggregation Consortium (Broad Institute) and NHLBI Exome Sequencing Project). The leucine295 position is highly conserved among species through Caenorhabditis elegans (Figure 2b). The ER lumenal MBOAT portion (244–488 amino acids) of this enzyme is the active domain that includes histidine 415 as the active site (Figure 2c). Additional in silico analysis of the protein stability using all available tools, including MuStab (prediction confidence 92.32%) and MUpro (confidence score −1), predicted decreased stability of the mutated protein.16 In addition, Leucine at the 295th position is located in the alpha helix region.17 Although Proline is a cyclic amino acid that is usually located at the beginning of a helix (one of the first three amino acids) in order to help the helix to bend, the Leucine-to-Proline change inside the helix causes a loss of hydrogen bond that can disturb the helix and profoundly affect the structure of the protein, and thus impair DGAT1 enzyme activity (Figure 2d).

Mutations in DGAT1 identified in our patients. (a) Chromatogram showing proband (IV-5) with homozygous mutation, c.884T>C (p.Leu295Pro). Parents (III.1 and III.2 – number in figure) and two of the unaffected siblings (IV-2 and IV-3) were heterozygous, and one of the unaffected brothers (IV-1) was normal. (b) Conservation of leucine at position 295, among different species across evolution. (c) Diagrammatic representation of Dgat1 protein domains, showing the sites of mutations and the catalytic domain of enzyme with active-site histidine415. The 25 amino acid in-frame deletion reported by Haas et al. and in our study (Family 2), in the highly conserved catalytic lumenal domain is also shown. (d) In silico protein modeling showing the loss of a hydrogen bond in the mutated protein that probably creates a hinge in the alpha helical region.
Exome sequencing in Family 2
Of 68 million 100-base reads obtained for the six samples (Supplementary Table 1), ~98% were successfully aligned to the reference human genome. Approximately 1000 non-common variants (max allele frequency smaller than 0.01) affecting protein sequence were detected in each sample. Using in-house scripts to extract only homozygous variants in the proband that demonstrated recessive inheritance and were not homozygous in the healthy siblings, we found five variants (Supplementary Table 2) in chromosomes 1 and 8. These regions demonstrate IBD2 allele sharing16 between the patients, but not with one of the healthy siblings (Supplementary Figure 1). One variant (RECQL4) was evident in our in-house sequence database with an allele frequency of 5%, and was excluded. The DGAT1 splicing mutation NG_034192.1:g.13827T>C, homozygous in both affected siblings (2-III-12 and 2-III-13) was present in both ClinVar and HGMD databases (rs148665132). This splicing mutation causes skipping of whole exon 8 of DGAT1 that result in the in-frame deletion of 25 amino acids (p.Ala226_Arg250del) in the protein level.4 Both parents, as well as two of the six unaffected siblings (male and female), were heterozygous for this mutation.
DGAT1 expression fibroblasts
DGAT1 is expressed in numerous human adult tissues and tissue culture cell lines with moderate expression in the liver, skeletal muscle, stomach, and predominant expression in the small intestine, adrenal gland, and testes. In our study, the novel variant in DGAT1 is related to diarrhea; therefore, it was ideal to study its expression in small intestinal cell lines. As these cell lines were unavailable from the patient and DGAT1 is reported to be moderately expressed in the skin, we measured the expression in skin fibroblasts in comparison with control to find out the effect of variant in gene expression. Analysis of mRNA from skin fibroblasts of patient IV-5 (Family 1) showed decrease in the level of DGAT1 expression (NM_012079) to ~65-70% compared to control (Figure 3a). Prediction of the effect of the variant using Human Splicing Finder (http://www.umd.be/HSF3/) revealed that it may create some Exon Splicing Enhancer and Silencer that could lead to partial splicing aberrations, potentially leading to nonsense mediated decay explaining this decrease. Analysis of the protein level using western blot (Figure 3b) also confirmed this reduction (~50% of control fibroblast).

DGAT1 expression is reduced in fibroblasts of Family 1 with a novel mutation in DGAT1. (a) qPCR of mRNA from control and patient (Family 1, IV-5) show reduced (65–70%) expression of DGAT1 in patient (black bar) relative to control (gray bar). Results were normalized with GAPDH expression. Error bars represent SD from six replicates. Asterisk (*) indicate a significance of P<0.05. (b) Cytoplasmic fraction of fibroblasts from control and patient (Family 1, IV-5) electrophoresed and transferred into nitrocellulose blot. Probing with DGAT1 antibody showed a reduced amount of protein in patient (gray bar) as compared with control (black bar). Protein loading was normalized with β-actin levels.
Frequency of the DGAT1 mutations in databases
We listed in Supplementary Table 3 all the missense and potential loss-of-function (Stop gain, frameshift, and splice variants) variants in DGAT1 reported in normal individuals from the Exome Aggregation Consortium of the Broad Institute (http://exac.broadinstitute.org) and tested the potential pathogenicity of the missense variants using PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org). We estimated the prevalence of DGAT1 deficiency by combining the allele frequencies of all the loss-of-function variants, and all missense mutations predicted to be deleterious by both PolyPhen2 (Probably deleterious) and SIFT (Damaging) to ~1/300 000 (Table 2). This translates to a frequency similar to that of a rare disease, but, as to date, only one family has been described;4 thus, DGAT1 deficiency is an underdiagnosed disease.
Discussion
Several reports describe variants in DGAT1 associated with altered milk production and fat content in cattle.18, 19 However, in humans only a single family has been reported associated with DGAT1 mutations. Haas et al described a non-consanguineous Ashkenazi-Jewish family in which two of three siblings were homozygous for a splice site mutation leading to exon 8 deletion associated with PLE, hypoalbuminemia, and early-onset diarrhea.4 This is the second report of patients with DGAT1 mutations.
All known DGAT1-deficient patients have manifested with early-onset non-bloody watery diarrhea, unresponsiveness to soy-based or elemental formula, and had clinical, laboratory, and pathological findings consistent with PLE. FTT, hypogammaglobulinemia with recurrent infections, and the need for albumin infusions and parenteral nutrition were common to the three reported families. Table 3 summarizes the clinical and molecular data regarding all patients with DGAT1-associated PLE. Several differences, however, are apparent among our reported cases. First, the serum lipid profile, including triglycerides, was normal in patient 1-IV-4, while mildly elevated in 1-IV-5, whereas patient 2-III-13 and the siblings reported by Haas et al.4 had elevated serum triglycerides. A larger study involving more patients to give more robust conclusions if hypertriglyceridemia is related to the patient’s genotype would likely be required. Second, both of our patients 1-IV-4 and 1-IV-5 had digital clubbing, not previously described. Third, the onset of diarrhea in our Family 1 was significantly later (2–4 months) than in the previously described cases (3 days) or in patients 2-III-12 and 2-III-13 of Family 2.
These phenotypic differences may reflect genotypic differences. While the previously reported family and our Family 2 patients showed a splice site mutation causing deletion of exon 8 and a null allele, our Family 1 patients harbored a novel missense variant, p.(Leu295Pro). Although the c.884T>C variant in DGAT1 is highly likely to cause our patient’s disease, absolute confirmation would require identification of similarly affected patients with this variant or the implementation of a highly specialized enzymatic assay of DGAT1 activity in a suitable cell system. Defective DGAT1 likely causes substrates to accumulate in intestinal cells, resulting in malabsorption and diarrhea; this effect is likely to be less severe when residual enzyme activity is present (as in a missense mutation) than when a null mutation produces no enzyme at all.
Interestingly, the previous family with DGAT1 mutations was not the first multiplex family to be reported with congenital PLE. A previous report from our own institution described a consanguineous family of Christian-Arab descent from Nazareth, Israel, of which 8 of 28 children born to two sibships were affected with growth retardation, edema, and growth restriction attributed to PLE, as well as digital clubbing.20 Although their clinical and pathological data were given, genetic information is lacking; however, these individuals may harbor mutations in DGAT1.
Although Dgat1 deficiency in mice has conferred resistance to diet-induced obesity and increased insulin and leptin sensitivity,7, 8, 21 this may well be different in humans. The susceptibility of humans, as compared with other mammals, on DGAT1 deficiency may be due to the expression of DGAT enzymes. Unlike in mice and other mammals where Dgat1 and Dgat2 are ubiquitously expressed, in humans DGAT1 is highly expressed in the intestines, while DGAT2 is not expressed at all.5, 7, 22 Thus, DGAT1 inhibition in patients, especially in the intestines, could cause unwanted symptoms. Of note, DGAT1 inhibitors have been proposed to treat obesity and type II diabetes mellitus.22 Interestingly, these inhibitors had dose-related adverse events including mild to moderate diarrhea,4 consistent with the DGAT1-deficient phenotype. As PLE and CDD condition-associated DGAT1 mutations appear to be underdiagnosed, the use of DGAT1-inhibitory drugs in metabolic diseases should be an approach to be taken with careful consideration.
Our findings expand the differential diagnosis of congenital diarrhea disorders that present in infancy with PLE. Clinicians should maintain a high index of suspicion for DGAT1 deficiency for cases with PLE, especially when consanguinity is apparent or when more than one family member is affected.
References
Umar SB, DiBaise JK : Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol 2010; 105: 43–49, quiz 50.
Lee YT, Sung JJ : Protein-losing enteropathy. Gastrointest Endosc 2004; 60: 801–802.
Canani RB, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G : Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastr Nutr 2010; 50: 360–366.
Haas JT, Winter HS, Lim E et al: DGAT1 mutation is linked to a congenital diarrheal disorder. J Clin Invest 2012; 122: 4680–4684.
Cases S, Smith SJ, Zheng YW et al: Identification of a gene encoding an acyl CoA: diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci USA 1998; 95: 13018–13023.
Cheng D, Iqbal J, Devenny J et al: Acylation of acylglycerols by acyl coenzyme a:diacylglycerol acyltransferase 1 (DGAT1) functional importance of DGAT1 in the intestinal fat absorption. J Biol Chem 2008; 283: 29802–29811.
Smith SJ, Cases S, Jensen DR et al: Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet 2000; 25: 87–90.
Chen HC, Smith SJ, Ladha Z et al: Increased insulin and leptin sensitivity in mice lacking acyl CoA: diacylglycerol acyltransferase 1. J Clin Invest 2002; 109: 1049–1055.
Wierenga KJ, Jiang ZJ, Yang AC, Mulvihill JJ, Tsinoremas NF : A clinical evaluation tool for SNP arrays, especially for autosomal recessive conditions in offspring of consanguineous parents. Genet Med 2013; 15: 354–360.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Li MX, Gui HS, Kwan JS, Bao SY, Sham PC : A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res 2012; 40: e53.
Livak K : Comparative Ct method. ABI Prism 7700 Sequence Detection System 1997.
Livak KJ, Schmittgen TD : Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K : REAP: a two minute cell fractionation method. BMC Res Notes 2010; 3: 294.
Cheng J, Randall A, Baldi P : Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006; 62: 1125–1132.
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G : Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11: 548.
Lehnert K, Ward H, Berry SD et al: Phenotypic population screen identifies a new mutation in bovine DGAT1 responsible for unsaturated milk fat. Sci Rep 2015; 5: 8484.
Lu J, Boeren S, van Hooijdonk T, Vervoort J, Hettinga K : Effect of the DGAT1 K232A genotype of dairy cows on the milk metabolome and proteome. J Dairy Sci 2015; 98: 3460–3469.
Shani M, Theodor E, Frand M, Goldman B : A family with protein-losing enteropathy. Gastroenterology 1974; 66: 433–445.
Chen HC : Enhancing energy and glucose metabolism by disrupting triglyceride synthesis: Lessons from mice lacking DGAT1. Nutr Metab 2006; 3: 10.
Cases S, Stone SJ, Zhou P et al: Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem 2001; 276: 38870–38876.
Acknowledgements
This research was supported by the Intramural Research Programs of the National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA; the I-CORE program (grants No. 41/11); and the Gene and Protein Expression core of the NIH-supported Cincinnati Children’s Hospital Research Foundation Digestive Health Center (1P30DK078392-01). We thank the patients and their families for their kind assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Stephen, J., Vilboux, T., Haberman, Y. et al. Congenital protein losing enteropathy: an inborn error of lipid metabolism due to DGAT1 mutations. Eur J Hum Genet 24, 1268–1273 (2016). https://doi.org/10.1038/ejhg.2016.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.5
This article is cited by
-
Beyond IBD: the genetics of other early-onset diarrhoeal disorders
Human Genetics (2023)
-
Diet and Management of Diarrhea
Indian Journal of Pediatrics (2023)
-
The genetics of monogenic intestinal epithelial disorders
Human Genetics (2023)
-
Apoptotic enteropathy, gluten intolerance, and IBD-like inflammation associated with lipotoxicity in DGAT1 deficiency–related diarrhea: a case report of a 17-year-old patient and literature review
Virchows Archiv (2022)
-
Overview of Albumin Physiology and its Role in Pediatric Diseases
Current Gastroenterology Reports (2021)

{kind=link}