
Abstract
Aim:
To determine whether ginsenosides with various sugar attachments may act as active components responsible for the cardiac therapeutic effects of ginseng and sanqi (the roots of Panax ginseng and Panax notoginseng) via the same molecular mechanism triggered by cardiac glycosides, such as ouabain and digoxin.
Methods:
The structural similarity between ginsenosides and ouabain was analyzed. The inhibitory potency of ginsenosides and ouabain on Na+/K+-ATPase activity was examined and compared. Molecular modeling was exhibited for the docking of ginsenosides to Na+/K+-ATPase.
Results:
Ginsenosides with sugar moieties attached only to the C-3 position of the steroid-like structure, equivalent to the sugar position in cardiac glycosides, and possessed inhibitory potency on Na+/K+-ATPase activity. However, their inhibitory potency was significantly reduced or completely abolished when a monosaccharide was linked to the C-6 or C-20 position of the steroid-like structure; replacement of the monosaccharide with a disaccharide molecule at either of these positions caused the disappearance of the inhibitory potency. Molecular modeling and docking confirmed that the difference in Na+/K+-ATPase inhibitory potency among ginsenosides was due to the steric hindrance of sugar attachment at the C-6 and C-20 positions of the steroid-like structure.
Conclusion:
The cardiac therapeutic effects of ginseng and sanqi should be at least partly attributed to the effective inhibition of Na+/K+-ATPase by their metabolized ginsenosides with sugar moieties attached only to the C-3 position of the steroid-like structure.
Similar content being viewed by others

Introduction
Ginseng and sanqi (the roots of Panax ginseng and Panax notoginseng), 2 well-known traditional Chinese medicinal herbs, have been greatly used in several Asian countries for thousands of years1. Despite certain distinct remedial usages, comparable therapeutic effects, such as the promotion of blood circulation, have been reported for these 2 herbs. Both ginseng and sanqi are commonly used for the treatment of coronary heart disease and cerebral vascular disease2, 3, 4. Belonging to the same genus, ginseng and sanqi possess similar constituents, including their unique active ingredients, ginsenosides5, 6. Cumulated research outcomes have indicated that identical and variable ginsenosides in these 2 herbs are possibly responsible for the comparable and distinct therapeutic effects between ginseng and sanqi7.
Ginsenosides are triterpene saponins that have a common 4 ring hydrophobic steroid-like structure with sugar moieties attached mostly at the C-3, C-6, or C-20 position8. So far, more than 80 ginsenosides have been isolated from over 10 Panax taxa, and most of these are derived from 4 types of aglycones: protopanaxadiol, protopanaxatriol, oleanolic acid, and ocotillol9. Biological activities, such as neuroprotective effects, antitumoral activity, and cardiac therapeutic effects have been documented for these ginsenosides1, 7, 10. The different sugar moieties in ginsenosides are assumed to provide specificity for the diverse therapeutic effects of variable ginsenosides. Although the cardiac therapeutic effects of ginsenosides have been documented, the detailed molecular mechanism triggering this medicinal role is still unknown. It has been speculated that calcium channel blocking or the antifree-radical action of ginsenosides may be responsible for their cardiac therapeutic effect11.
Cardiac glycosides, such as ouabain and digoxin, also contain a core steroid-like structure and have been utilized for the treatment of congestive heart failure12. The cardiac therapeutic effect of cardiac glycosides is a result of their reversible inhibition on the α-subunit of the membrane-bound Na+/K+-ATPase, mainly, but not exclusively, located in the human myocardium13. This inhibition leads to the accumulation of sodium in cardiac cells, which are enforced to promote the sodium-calcium exchange system in the cell membrane, thus causing a higher level of intracellular and myocardial calcium. The elevated intracellular calcium induces positive inotropy that eventually accentuates the force of myocardial contraction.
In light of the structural similarity between ginsenosides and cardiac glycosides, as well as their consentaneous utilization in cardiac therapy, we determined whether ginsenosides of certain sugar attachments may act as active ingredients responsible for the cardiac therapeutic effects of ginseng and sanqi via the same molecular mechanism triggered by cardiac glycosides. In this study, the inhibitory potency of various ginsenosides and ouabain on Na+/K+-ATPase was examined and compared. The molecular modeling and docking of representative ginsenosides to Na+/K+-ATPase were exhibited to reveal the observed difference in Na+/K+-ATPase inhibition among ginsenosides at the molecular level.
Materials and methods
Chemicals and reagents
Ginsenosides Rb1, Rb2, Rb3, Rc, Rd, Rg3, Rh2, PPD, Re, Rf, Rg2, Rg1, Rh1, and PPT, as well as pseudoginsenoside F11, were purchased from Scientific Pharmaceutical Elite Company (Taiwan). Ginsenoside R1 was obtained from ChromaDex (Irvine, CA, USA). Oleanolic acid and ouabain were obtained from Sigma (St Louis, MO, USA). The phosphate assay kit was purchased from Amresco (Solon, Ohio, USA).
Measurement of Na+/K+-ATPase activity
The Na+/K+-ATPase activity was determined by measuring the amount of inorganic phosphate (Pi) liberated from ATP14. A commercial Na+/K+-ATPase from the porcine cerebral cortex (Sigma, USA; 0.3 units/mg) was incorporated into a 1 mL reaction mixture containing 3 mmol/L ATP, 5 mmol/L MgCl2, 80 mmol/L NaCl, 20 mmol/L KCl, and 40 mmol/L Tris-HCl (pH 7.8). The enzymatic reaction was terminated by adding 250 μL of 30% (w/v) trichloroacetic acid after the incubation period. After centrifugation at 10 000×g for 10 min, the supernatant was diluted 12.5-fold with deionized water and then 50 μL color development reagent, which was provided by the phosphate assay kit, was added. After 30 min of incubation at room temperature, the color intensity was measured at 620 nm on a SpectraMax M2 reader (Molecular Devices, Sunnyvale, CA, USA). Sodium pump activity was expressed as μmol Pi liberated from ATP by 1 mg of Na+/K+-ATPase in 1 h.
Data analysis
Data were expressed as mean±SEM of 5 replicates, and one-way ANOVA was performed on SPSS 12.0 for Windows (SPSS, Chicago, IL, USA). Differences were considered statistically significant at P<0.05.
Molecular modeling and docking
The crystal structure of pig renal Na+/K+-ATPase (Protein Data Bank [PDB] code 3B8E) was downloaded from the PDB15. In order to facilitate the docking process, we removed the β and γ subunits of the Na+/K+-ATPase, as well as the water molecules and counter ions surrounding the remaining α subunit. The modified Na+/K+-ATPase after hydrogen saturation was minimized with the CHARMm force field16 using the Discover Studio 2.0 package (http://accelrys.com/products/discovery-studio). The 2-D structures of ginsenosides used in this study were constructed by using the ChemDraw program, and their corresponding 3-D structures were converted by the Chem3D program (http://www.cambridgesoft.com). All of the ginsenosides were either 20(S)-protopanaxadiol or 20(S)-protopanaxatriol. The pocket for binding cardiac glycosides in the Na+/K+-ATPase α subunit was defined among the extracellular loops linking transmembrane segments as reported by Qiu et al17, 18, and the subunit-binding domain was defined as the region of the sphere with a 10 Å radius from the center of the binding pocket, which lies between I315 and L793 of the modified Na+/K+-ATPase. Docking of the ginsenosides was performed in silico by employing the LibDock module19 in the Discover Studio 2.0 package. There were 100 hotspots identified in the binding pocket. The LibDock methodology effectively executed the docking of combinatorial libraries of compounds in a high-throughput manner while keeping the protein structure fixed20. With the energy threshold set as 20 kcal/mol, 76 conformations of Rg3, 118 conformations of Rh2, 26 conformations of PPD, and 25 conformations of PPT were generated for docking, respectively. The ginsenoside-Na+/K+-ATPase complexes were evaluated by LibDockscore21. This scoring resembles piecewise linear potential22 by summing over interacting atoms in the specific ginsenoside-Na+/K+-ATPase complexes.
Results
Structural comparison between ginsenosides and ouabain
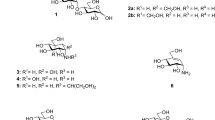
Similar to cardiac glycosides, such as ouabain, ginsenosides comprise a core steroid-like structure with sugars and other functional groups attached at variable positions (Figure 1). Protopanaxadiol ginsenosides, 1 of the 2 major ginsenoside groups, possess sugar moieties at the C-3 and C-20 positions, while protopanaxatriol ginsenosides, the other major group, possess a hydroxyl group at the C-3 position and sugar moieties at the C-6 and C-20 positions of the steroid-like structure. Among the 3 sugar attachment positions in ginsenosides, the C-3 position of the steroid-like structure is equivalent to the unique sugar attachment position of cardiac glycosides.

Chemical structures of 7 protopanaxadiol ginsenosides and their aglycone PPD, 6 protopanaxatriol ginsenosides and their aglycone PPT, pseudoginsenoside F11, oleanolic acid, and ouabain.
Inhibition of porcine Na+/K+-ATPase by ginsenosides
To verify whether ginsenosides and their aglycones exhibit a similar therapeutic effect via the same mechanism triggered by ouabain, that is, accentuating the force of myocardial contraction by elevating calcium concentration via the inhibition of Na+/K+-ATPase, a commercial Na+/K+-ATPase from the porcine cerebral cortex was used to evaluate the inhibitory potency of 7 protopanaxadiol ginsenosides and their aglycone PPD, 6 protopanaxatriol ginsenosides and their aglycone PPT, pseudoginsenoside F11, oleanolic acid, and ouabain. Among the protopanaxadiol ginsenosides, Rg3, Rh2, and PPD, which contain no sugar moieties at the C-20 position of the steroid-like structure, showed significant inhibition on Na+/K+-ATPase activity, regardless of the sugar attachment at the C-3 position; however, the inhibitory potency was mostly reduced (for ginsenoside Rd) or completely abolished (for ginsenosides, Rb1, Rb2, Rb3, and Rc) when monosaccharide or disaccharide was attached to the C-20 position (Figure 2A). All of the 6 protopanaxatriol ginsenosides (Re, R1, Rf, Rg1, Rg2, and Rh1) containing sugar moieties at the C-6 position of the steroid-like structure did not inhibit Na+/K+-ATPase, while PPT, which does not contain sugar moieties at the C-6 and C-20 positions, possessed considerable inhibitory potency (Figure 2B). Similar to protopanaxatriol ginsenosides, pseudoginsenoside F11, which contains a disaccharide attached at the C-6 position, did not inhibit Na+/K+-ATPase, while oleanolic acid, which does not contain sugar, possessed significant inhibitory potency (Figure 2C). Under our experimental conditions, the inhibitory potency of all ginsenosides on porcine Na+/K+-ATPase was evidently lower than that of ouabain; the IC50 of ouabain (0.45 μmol/L) was approximately 120 times lower than that of ginsenoside Rh2 (55 μmol/L), the strongest inhibitor of Na+/K+-ATPase among the ginsenosides examined in this study (Figure 3).

Inhibition of porcine Na+/K+-ATPase by ginsenosides. Inhibitory potency of (A) 7 protopanaxadiol ginsenosides and their aglycone PPD, (B) 6 protopanaxatriol ginsenosides and their aglycone PPT, and (C) pseudoginsenoside F11 (PF11), oleanolic acid (OA), and ouabain (OUA) were observed as the reduction of Pi liberation released from ATP by a constant amount of commercial porcine Na+/K+-ATPase. Data represent mean±SEM of 5 replicates. bP<0.05, cP<0.01 vs control group (CON; deionized water only).

Inhibitory potency of ouabain and ginsenoside Rh2 on porcine Na+/K+-ATPase. Inhibitory potency of various concentrations of ouabain and ginsenoside Rh2 was observed as the reduction of Pi liberation released from ATP by a constant amount of commercial porcine Na+/K+-ATPase.
Molecular modeling and docking of ginsenosides to Na+/K+-ATPase
To reveal the observed difference in Na+/K+-ATPase inhibition among ginsenosides at the molecular level, representative ginsenosides, Rg2, Rg3, Rd, Rb1, PPD, PPT, and Rh1 were subjected to molecular modeling and docking to the extracellular domain of the α subunit of Na+/K+-ATPase. In accordance with the results of the Na+/K+-ATPase inhibitory assay for these representative ginsenosides, only PPD, Rh2, and Rg3, which contained 0, 1, and 2 sugar units at the C-3 position, but no sugar attached at the C-6 and C-20 positions, could dock into the extracellular pocket of the Na+/K+-ATPase α subunit (Figure 4). The core steroid-like structure of PPD, Rh2, or Rg3 was trapped in a cave (shown in green with dimensional limitations of 10.5 and 9.2 Å for the top and bottom areas, respectively) of the extracellular pocket. The protrusive alkyl side chain attached at the C-20 position of the steroid-like structure was located in a narrow space deeper than the cave. In comparison with PPD, the extra monosaccharide and disaccharide of Rh2 and Rg3 at the C-3 position of the steroid-like structure were located outside of the cave and were freely exposed to the extracellular space.

Modeling of ginsenosides binding to the extracellular pocket of the Na+/K+-ATPase α subunit. (A) core steroid-like structure of ginsenoside Rg3 was trapped in a cave (shown in green with dimensional limitations of 10.5 and 9.2 Å for the top and bottom areas, respectively) of the extracellular pocket of the Na+/K+-ATPase α subunit. Similar structural interaction was observed for ginsenoside Rh2 (Rg3 without the orange glucose molecule) and PPD (Rg3 without the orange and pink glucose molecules). (B) molecular interaction between ginsenoside Rg3 and the extracellular binding pocket of the Na+/K+-ATPase α subunit. Ginsenoside Rg3 and the binding pocket of Na+/K+-ATPase were displayed in ball-and-stick and space-fill modes, respectively. Left-side view (C) of the extracellular space facing the binding pocket of Na+/K+-ATPase (B).
Detailed molecular interactions between Rg3, Rh2, PPD, or PPT and the extracellular pocket of Na+/K+-ATPase are shown in Figure 5. Three hydrogen bonds were found between the sugar moieties at the C-3 position of Rg3 or Rh2 and Na+/K+-ATPase (Figure 5A, 5B). Among these hydrogen bond interactions, R880 of Na+/K+-ATPase was involved in 2 hydrogen bonds with the sugar moieties at the C-3 position of Rh2. Presumably, the interaction between the sugar moieties at the C-3 position of ginsenosides was beneficial for their binding to the extracellular pocket of Na+/K+-ATPase. Only 1 hydrogen bond was observed between the hydroxyl group at the C-20 position of PPD and V798 of Na+/K+-ATPase (Figure 5C). Two hydrogen bonds were observed between PPT and Na+/K+-ATPase; one was between the hydroxyl group at the C-3 position of PPT and N790 of Na+/K+-ATPase, and the other between the hydroxyl group at the C-12 position of PPT and R904 of Na+/K+-ATPase (Figure 5D). Our observation of hydrogen bond interactions between Arg and ginsenosides was in accordance with that between Arg in the H7–H8 loop of human Na+/K+-ATPase and cardiac glycosides23. The interaction between ginsenosides and the extracellular pocket of Na+/K+-ATPase putatively blocked the entrance of ions rather than impeded the active site of Na+/K+-ATPase located in the intracellular region. LibDockscores of the Rg3, Rh2, PPD, and PPT were 145.069, 148.118, 100.344, and 103.058, respectively. The docking score results were consistent with the observations in the inhibitory assay.

Detailed molecular interactions between the extracellular pocket of Na+/K+-ATPase and (A) Rg3, (B) Rh2, (C) PPD, and (D) PPT. Amino acid residues of Na+/K+-ATPase close to ginsenosides are shown in the wire frame, and the structures of ginsenosides are shown in the scaled ball and stick. Green dashed lines represent the hydrogen bonds between ginsenosides and Na+/K+-ATPase. Amino acids involved in hydrogen bonding to ginsenosides are in green.
To rationalize the steric hindrance of sugar attachment at the C-6 or C-20 position of the steroid-like structure, the molecular fitness of PPD, PPT, and Rh1 or Rg3, Rd, and Rb1 in the cave of the extracellular pocket of the Na+/K+-ATPase α subunit was depicted and compared (Figure 6). The altered dimension of ginsenoside increased from 6.1 Å (PPD) to 7.1 Å (PPT) or 11.8 Å (Rh1) when a hydroxyl group or glucose was attached to the C-6 position of the steroid-like structure (Figure 6, left). Since the dimensional limitations of the extracellular cave of the Na+/K+-ATPase α subunit were 10.5 and 9.2 Å, both PPD and PPT, but not Rh1, could fit in the cave. Therefore, PPD and PPT, but not Rh1, possessed inhibitory potency on Na+/K+-ATPase. The altered dimension of ginsenoside increased from 3.1 Å (Rg3) to 9.6 Å (Rd) or 14.6 Å (Rb1) when monosaccharide or disaccharide was attached to the C-20 position of the steroid-like structure (Figure 6, right). Consequently, Rg3, but not Rb1, could fit in the cave and possessed inhibitory potency on Na+/K+-ATPase. Rd (9.6 Å) could pass through the top (10.5 Å), but could not reach the bottom (9.2 Å) of the cave, and thus possessed subtle inhibitory potency on Na+/K+-ATPase.

Molecular fitness of representative ginsenosides in the cave of the extracellular binding pocket of the Na+/K+-ATPase α subunit. Green cave of dimensional limitations for ginsenoside fitness was the same as that shown in Figure 4. Red and brown colors show the glucose molecules attached to the C-6 and C-20 positions of the steroid-like structure. Yellow balls indicate the extended dimensions of ginsenosides after hydroxylation (PPT) or glycosylation (Rh1, Rd, and Rb1) at the C-6 or C-20 positions.
Discussion
Based on experimental observation and theoretical modeling, we found that ginsenosides with sugar moieties attached only to the C-3 position of the steroid-like structure, equivalent to the sugar position in cardiac glycosides, and possessed inhibitory potency on Na+/K+-ATPase activity. Sugar attachment to the C-6 or C-20 position of the steroid-like structure presumably caused steric hindrance for the entrance of ginsenosides into the extracellular binding pocket of the Na+/K+-ATPase α subunit, and thus significantly reduced or completely abolished their inhibitory potency. Paradoxically, most ginsenosides found in ginseng and sanqi have not appeared to be competent inhibitors for Na+/K+-ATPase due to their sugar attachment to the C-6 or C-20 position of the steroid-like structure. Nevertheless, ginsenosides might act as prodrugs, as they tend to be metabolized to their active forms by intestinal bacterial deglycosylation after oral administration8. Commonly, the metabolites could be easily absorbed by the intestines due to the increase of hydrophobicity after deglycosylation, and might display the same or different pharmacological actions in comparison with their parent compounds24, 25. While our experimental observation and theoretical modeling were executed using porcine Na+/K+-ATPase, the observed inhibitory potency of ginsenosides was presumably applicable to human Na+/K+-ATPase, since isoforms of this enzyme in diverse species were highly conserved evolutionarily. In fact, the amino acid sequence identity between porcine and the human α1 subunit of Na+/K+-ATPase was found to be up to 98%. In view of the results observed in the current study and those reported in the available literature, we propose that the cardiac therapeutic effects of ginseng and sanqi should be at least partly attributed to the effective inhibition of Na+/K+-ATPase by their metabolized ginsenosides, with sugar moieties attached only to the C-3 position of the steroid-like structure.
Ginsenosides are also demonstrated to be the pharmacologically-active ingredients responsible for the effects of ginseng on the central and peripheral nervous systems7. They have been reported to possess reversible and selective inhibitory effects on voltage-dependent ion channels (such as the Ca2+, K+, and Na+ channels) and ligand-gated ion channels (such as N-methyl-D-aspartate, some subtypes of nicotinic acetylcholine, and 5-hydroxytryptamine type 3 receptors), although little is known about the exact mechanisms. As the inhibition of Na+/K+-ATPase also leads to the fluctuation of Ca2+, K+, and Na+ concentrations, it will be interesting to see if any cross-talks are held among these ion channels after being inhibited by ginsenosides, and as a result, exhibit the pharmacological actions of ginseng and sanqi.
Cardiac glycosides have been demonstrated to provide neuroprotection against ischemic stroke in a cortical brain slice-based compound screening platform26. Neuroprotective activity and delayed therapeutic potential were observed for cardiac glycosides in this brain slice assay model. Recently, we demonstrated that magnesium lithospermate B, a derivative of caffeic acid tetramer, present as the major soluble ingredient in danshen (the dried roots of medicinal plant Salvia miltiorrhiza), possessed a cardiac therapeutic activity by its effective inhibition on Na+/K+-ATPase and exhibited the same neuroprotective effect against ischemic stroke in a similar brain slice assay model27. Similar to cardiac glycosides and magnesium lithospermate B, we found that ginsenosides possessed inhibitory potency on Na+/K+-ATPase in this study. In previously published studies, similar neuroprotective effects were reported for ginsenosides against ischemic stroke, and some of the results were observed under the same brain slice assay model28, 29, 30, 31, 32. It remains to be investigated whether ginsenosides, as well as cardiac glycosides and magnesium lithospermate B, exert neuroprotection against ischemic stroke via the same mechanism triggered by the inhibition of Na+/K+-ATPase.
Author contribution
Jason TC TZEN designed research; Ronald JY CHEN and Tse-yu CHUNG performed research; Feng-yin LI and Nan-hei LIN contributed new analytical tools and reagents; Jason TC TZEN wrote the paper.
References
Sengupta S, Toh SA, Sellers LA, Skepper JN, Koolwijk P, Leung HW, et al. Modulating angiogenesis: the yin and the yang in ginseng. Circulation 2004; 110: 1219–25.
Lei XL, Chiou GC . Cardiovascular pharmacology of Panax notoginseng (Burk) F. H. Chen and Salvia miltiorrhiza. Am J Chin Med 1986; 14: 145–52.
Wei JX, Du YC . Modern science research and application of Panax notoginseng. Kunming: Yunnan Science and Technology Press;1996 (in Chinese).
Cicero AF, Vitale G, Savino G, Arlett R . Panax notoginseng (Burk.) effects on fibrinogen and lipid plasma level in rats fed on a high-fat diet. Phytother Res 2003; 17: 174–8.
Dong TT, Cui XM, Song ZH, Zhao KJ, Ji ZN, Lo CK, et al. Chemical assessment of roots of Panax notoginseng in China: regional and seasonal variations in its active constituents. J Agric Food Chem 2003; 51: 4617–23.
Leung KS, Chan K, Bensoussan A, Munroe MJ . Application of atmospheric pressure chemical ionisation mass spectrometry in the identification and differentiation of Panax species. Phytochem Anal 2007; 18: 146–50.
Hasegawa H . Proof of the mysterious efficacy of ginseng: basic and clinical trials: metabolic activation of ginsenoside: deglycosylation by intestinal bacteria and esterification with fatty acid. J Pharmacol Sci 2004; 95: 153–7.
Nah SY, Kim DH, Rhim H . Ginsenosides: are any of them candidates for drugs acting on the central nervous system? CNS Drug Rev 2007; 13: 381–404.
Zhu S, Zou K, Cai S, Meselhy MR, Komatsu K . Simultaneous determination of triterpene saponins in ginseng drugs by high-performance liquid chromatography. Chem Pharm Bull 2004; 52: 995–8.
Dan M, Su M, Gao X, Zhao T, Zhao A, Xie G, et al. Metabolite profiling of Panax notoginseng using UPLC-ESI-MS. Phytochemistry 2008; 69: 2237–44.
Jiang Y, Liu W, Wang XM, Zhong GG, Zhang WJ, Chen L, et al. Calcium channel blockade and anti-free-radical actions of panaxatriol saponins in cultured myocardiocytes. Acta Pharmacol Sin 1996; 17: 138–41.
Li-Saw-Hee FL, Lip GY . Digoxin revisited. QJM 1998; 91: 259–64.
Lin SC, Way EL . A high affinity Ca2+-ATPase enriched nerve-ending plasma membranes. Brain Res 1982; 235: 387–92.
Goldberg H, Fernandez A . Simplified method of the estimation of inorganic phosphorus in body fluids. Clin Chem 1966; 12: 871–82.
Morth JP, Pedersen BP, Toustrup-Jensen MS, Sørensen TL, Petersen J, Andersen JP, et al. Crystal structure of the sodium-potassium pump. Nature 2007; 450: 1043–9.
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M . CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comp Chem 1983; 4: 187–217.
Liu LY, Koenderink JB, Swarts HG, Willems PH, De Pont JJ . Phe783, Thr797, and Asp804 in transmembrane hairpin M5-M6 of Na+,K+-ATPase play a key role in ouabain binding. J Biol Chem 2003; 278: 47240–4.
Qiu LY, Krieger E, Schaftenaar G, Swarts HG, Willems PH, De Pont JJ, et al. Reconstruction of the complete ouabain-binding pocket of Na,K-ATPase in gastric H, K-ATPase by substitution of only seven amino acids. J Biol Chem 2005; 280: 32 349–55.
Dixon SL, Merz KM Jr . One-dimensional molecular representations and similarity calculations: methodology and validation. J Med Chem 2001; 44: 3795–809.
Rao SN, Head MS, Kulkarni A, LaLonde JM . Validation studies of the site-directed docking program LibDock. J Chem Inf Model 2007; 47: 2159–71.
Diller DJ, Merz KM Jr . High throughput docking for library design and library prioritization. Proteins 2001; 43: 113–24.
Willett P, Barnard JM, Downs GM . Chemical similarity searching. J Chem Inf Comput Sci 1998; 38: 983–96.
Schultheis PJ, Wallick ET, Lingrel JB . Kinetic analysis of ouabain binding to native and mutated forms of Na, K-ATPase and identification of a new region involved in cardiac glycoside interactions. J Biol Chem 1993; 268: 22 686–94.
Kobashi K, Akao T . Relation of intestinal bacteria to pharmacological effects of glycosides. Biosci Microflora 1997; 16: 1–7.
Tawab MA, Bahr U, Karas M, Wurglics M, Schubert-Zsilavecz M . Degradation of ginsenosides in humans after oral administration. Drug Metab Dispos 2003; 31: 1065–71.
Wang JK, Portbury S, Thomas MB, Barney S, Ricca DJ, Morris DL, et al. Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proc Natl Acad Sci USA 2006; 103: 10461–6.
Tzen JT, Jinn TR, Chen YC, Li FY, Cheng FC, Shi LS, et al. Magnesium lithospermate B possesses inhibitory activity on Na+,K+-ATPase and neuroprotective effects against ischemic stroke. Acta Pharmacol Sin 2007; 28: 609–15.
Zhang YG, Liu TP . Influences of ginsenosides Rb1 and Rg1 on reversible focal brain ischemia in rats. Acta Pharmacol Sin 1996; 17: 44–8.
Tian J, Fu F, Geng M, Jiang Y, Yang J, Jiang W, et al. Neuroprotective effect of 20(S)-ginsenoside Rg3 on cerebral ischemia in rats. Neurosci Lett 2005; 374: 92–7.
Zhou XM, Cao YL, Dou DQ . Protective effect of ginsenoside-Re against cerebral ischemia/reperfusion damage in rats. Biol Pharm Bull 2006; 29: 2502–5.
Yuan QL, Yang CX, Xu P, Gao XQ, Deng L, Chen P, et al. Neuroprotective effects of ginsenoside Rb1 on transient cerebral ischemia in rats. Brain Res 2007; 1167: 1–12.
Chen LM, Zhou XM, Cao YL, Hu WX . Neuroprotection of ginsenoside Re in cerebral ischemia-reperfusion injury in rats. J Asian Nat Prod Res 2008; 10: 439–45.
Acknowledgements
Project supported by a grant to Jason TC TZEN from the National Science Council, Taiwan, China (No 96-2752-B-005-008-PAE).
We thank Prof Chih-Ning SUN (Department of Entomology, National Chuang Hsing University, Taichung, Taiwan, China) for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, R., Chung, Ty., Li, Fy. et al. Effect of sugar positions in ginsenosides and their inhibitory potency on Na+/K+-ATPase activity. Acta Pharmacol Sin 30, 61–69 (2009). https://doi.org/10.1038/aps.2008.6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2008.6
Keywords
This article is cited by
-
Transcriptome analysis of Panax zingiberensis identifies genes encoding oleanolic acid glucuronosyltransferase involved in the biosynthesis of oleanane-type ginsenosides
Planta (2019)
-
Rapid preparation of rare ginsenosides by acid transformation and their structure-activity relationships against cancer cells
Scientific Reports (2015)
-
The pseudoginsenoside F11 ameliorates cisplatin-induced nephrotoxicity without compromising its anti-tumor activity in vivo
Scientific Reports (2014)
-
Detecting metabolites of different transition metallithospermate B complexes after intravenous injection in rats
Acta Pharmacologica Sinica (2014)
-
Enhancing the potency of lithospermate B for inhibiting Na+/K+-ATPase activity by forming transition metal ion complexes
Acta Pharmacologica Sinica (2013)
