
Abstract
Recently, the International Consensus Classification (ICC) and the 5th edition of the World Health Organization classification (WHO2022) introduced diagnostically similar yet distinct approaches, which has resulted in practical confusion. This review compares these classification systems for acute myeloid leukemia (AML), building up on the revised 4th edition of WHO (WHO2016). Both classifications retain recurrent genetic abnormalities as a primary consideration. However, they differ in terms of blast threshold. The ICC mandates a minimum of 10% blasts in the bone marrow or peripheral blood, whereas the WHO2022 does not specify a blast cut-off. AML with BCR::ABL1 requires > 20% blast count in both classifications. In WHO2022, AML with CEBPA mutation requires > 20% blasts. TP53 mutation, a new entity is exclusive to ICC, diagnosed with > 20% blasts and variant allele frequency > 10%. AML with myelodysplasia-related changes is defined by cytogenetic or gene mutation-based criteria, not morphological dysplasia. Eight genes were common to both groups: ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, and ZRSR2. An additional gene, RUNX1, was included in the ICC classification. AML cases defined by differentiation (WHO2022) and AML not otherwise specified (ICC) are categorized as lacking specific defining genetic abnormalities, WHO2022 labels this as a myeloid neoplasm post cytotoxic therapy (MN-pCT), described as an appendix after specific diagnosis. Similarly, in ICC, it can be described as “therapy-related”, without a separate AML category.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The World Health Organization (WHO) classification of hematolymphoid tumors has long served as an international diagnostic criterion. However, in 2022, the International Consensus Classification (ICC) and the 5th edition of the WHO classification (WHO2022) offered similar but distinct diagnostic approaches, leading to confusion [1,2,3,4,5]. Since the French-American-British classification in 1976, subsequent updates like WHO2001, WHO2008, and WHO2016, have incorporated new diagnostic criteria that integrate molecular, pathological, and clinical variables into a morphological classification [6,7,8,9]. The myeloblast threshold in diagnostic criteria has gradually decreased, with genetic abnormalities emerging as a crucial criterion. The evolution has made personalized management more feasible over time. This review explores the changes from the revised 4th edition of WHO2016 to WHO 2022 and the ICC classification, focusing on acute myeloid leukemia (AML).
WHO2016: acute myeloid leukemia with recurrent genetic abnormalities
Genetic abnormalities continue to be key diagnostic criteria. The WHO2016 classification, which defined “AML with recurrent genetic abnormalities”, was renamed “AML with defining genetic abnormalities” in WHO2022 [10]. While maintaining the same ICC, additional “other rare recurring translocations” subgroups were created [5]. Both WHO2022 and ICC were broader in scope compared to WHO2016 (Table 1).
The key change in WHO2022 is the exclusion of the myeloblast percentage threshold for diagnosis when specific genetic abnormalities are present. Unlike WHO2016, where the myeloblast count was not a significant factor in diagnosing certain AML subtypes, such as AML with t(8;21)(q22;q22.1), AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22), and acute promyelocytic leukemia (APL) with PML-RARA, WHO2022 now applies myeloblast count criteria to additional genetic abnormalities such as t(9;11)(p21.3;q23.2), t(6;9)(p23;q34.1), inv(3)(q21.3q26.2), t(3;3)(q21.3;q26.2) or t(1;22)(p13.3;q13.1), while excluding AML with BCR::ABL1 fusion and CEBPA mutation. A novel structure for “AML with other defined genetic alterations” was introduced, including new and/or uncommon AML subtypes that may be included in future editions. The ICC further categorized subgroups into “AML with recurrent genetic abnormalities” and “other rare recurring translocations”. Notable differences include 1) the incorporation of additional RARA, KMT2A, and MECOME rearrangements and 2) a requirement for blast count exceeding 10% for diagnosis, except in cases of t(9;22)(22)(q34.1;q11.2)/BCR::ABL1 and TP53 mutations, which require a blast count exceeding 20%.
In WHO2016 classification, AML with mutated NPM1, AML with biallelic mutations in CEBPA, and AML with mutated RUNX1 were classified as AML with genetic mutations. AML with mutated NPM1 and CEBPA remained classified in both the WHO2022 and ICC classifications. However, AML with mutated RUNX1 was excluded from the provisional diagnosis due to its limited clinical significance.
AML with mutated NPM1
NPM1-mutated AML has been recognized as a distinct entity since 2008. Morphologically, blasts exhibit monocytic differentiation, and this subtype is frequently observed in young patients with a high prevalence of the normal karyotype [11,12,13]. There is a discrepancy in the blast threshold for diagnosis between the ICC and WHO2022. ICC requires a blast count ≥ 10%, whereas WHO2022 does not specify a blast number cutoff. Although an increase in blasts exists in most AML cases with mutated NPM1, if the blast count is < 10%, the diagnosis is changed to “AML with NPM1” in WHO2022 and “NPM1-mutated myelodysplastic syndrome (MDS)” in ICC. NPM1 mutations are also detected in MDS and MDS/MPN [14], occurring in approximately 2% of MDS, cases with excess blasts [15], leading to potential confusion in clinical management and treatment decisions.
Especially concerning AML with biallelic mutation of CEBPA
In WHO2022, this category is termed “AML with CEBPA mutation”, encompassing biallelic (biCEBPA) and single mutations in the basic leucine zipper region (smbZIP-CEBPA) [10]. Conversely, ICC designates the diagnosis as “AML with mutated bZIP CEBPA”, emphasizing the bZIP domain mutation irrespective of its mono or biallelic nature. This conclusion is supported by recent studies demonstrating that bZIP domain mutations are linked to favorable clinical outcomes [4, 16]. The blast count diagnostic criteria in ICC, consistent with other entities, is ≥ 10%. In contrast, WHO2022 suggests a blast count of ≥ 20%. Common morphological features often indicate AML with maturation (FAB M2) or AML without maturation (FAB M1) [17]. However, distinctive morphological features are lacking and occur at a frequency of 7–16% in adults and 4.5–15% in pediatric patients [4].
AML with TP53 mutation
Notably, TP53 was not included in the WHO2022 AML with defined genetic abnormalities. Instead, a biallelic TP53 alteration subtype is recognized in MDS, which is considered equivalent to AML. The diagnostic criteria for TP53 alterations in ICC require a blast count ≥ 20%, a higher threshold than in other entities, in conjunction with a variant allele frequency ≥ 10%. Therefore, when the blast count is < 20% in peripheral blood and bone marrow, MDS is characterized by both classifications. In WHO2022, “MDS with biallelic TP53 inactivation (MDS-biTP53)” is defined for cases with < 20% blasts, whereas ICC delineates “MDS mutated TP53” according to blast count differences. In addition, in cases of monoallelic loss, there was no significant clinical difference compared to the wild type [18]. Therefore, both classifications focus on biallelic loss.
AML with NUP98 rearrangement
This category is a newly introduced as “AML with NUP98 rearrangement” in WHO2022 and as “AML with t(5;11)(q35.2;p15.4)/NUP98::NSD1 and with t(11;12)(p15.4;p13.3)/NUP98:KMD5A and NUP98 and other partners” in ICC. NUP98 exhibits multiple fusion partners and, although infrequent, is associated with a poor prognosis [19]. The blast count requirement was maintained as a minimum for both classifications.
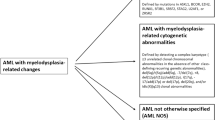
WHO2016: AML with myelodysplasia-related changes
In WHO2022, this category was named “AML with myelodysplasia-related (AML-MR)”, and ICC classified it as “AML with myelodysplasia-related gene mutation” and “AML with myelodysplasia- related cytogenetic abnormalities”. It was incorporated from an independent category into “AML with defining genetic abnormalities” in WHO2022. Both classification systems exclude morphology-based diagnostic criteria and emphasize molecular abnormalities. Some existing cytogenetic criteria have been updated, and gene mutations have been added. The myeloblast threshold requires ≥ 20% in both peripheral blood or bone marrow for this category.
AML with myelodysplasia-related cytogenetic abnormalities
Although there were no significant differences from the previous WHO2016, some distinctions were observed between the two classification systems (Table 2). Complex karyotype (≥ 3 abnormalities) and chromosomal aberrations on chromosomes 5, 7, 12, 17, and X were common in both systems. In the ICC, del(11q) was excluded, and +8 and del(20q) were added. Additionally, balanced abnormalities in WHO2016 were moved to “AML with other rare recurring translocations”.
AML with myelodysplasia-related gene mutations
Eight genes were common to both groups: ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, and ZRSR2. An additional gene, RUNX1, was included in ICC. Minimum variant allele frequencies are not required for these genes. They are associated with an adverse prognosis [20, 21].
WHO2016: therapy-related myeloid neoplasm
In WHO2022, a category named “Myeloid neoplasms post cytotoxic therapy (MN-pCT)” was introduced, encompassing AML, MDS, and MDS/MPN that develop after cytotoxic therapy inducing DNA damage [10]. Cytotoxic therapies, such as PARP1 inhibitors and methotrexate, were excluded. It is recommended to append “post cytotoxic therapy” after the specific diagnosis. Similarly, the ICC no longer recognized it as a distinct entity of AML.
WHO2016: acute myeloid leukemia, not otherwise specified
This group lacked genetic abnormalities and was classified based on morphology. Although this subtype has limited prognostic significance, it offers a practical paradigm [5, 10]. Both WHO and ICC maintain diagnostic criteria of ≥ 20% myeloblasts. Additionally, this category includes cases with overlapping phenotypic markers of the two lineages, such as mixed phenotype acute leukemia (MPAL) and early T-precursor lymphoblastic leukemia/lymphoma (ETT-ALL). Until recently, the genomics of MPAL has been predominantly associated with KMT2A rearrangement. However, recent findings have highlighted the involvement of the RAS pathway in B/M MPAL, the JAK/STAT pathway in T/M MPAL, ZEB2-BCL11B, NUP214-ABL1, and ETV6 in T/myeloid cells. These discoveries suggest the potential for future addition of new entities [10, 22].
European LeukemiaNet (ELN) risk stratification 2022
Aligned with the updated AML classification system that emphasizes genetic mutations, the ELN has released 2022 risk stratification guidelines based on the ICC classification (Table 3) [23, 24]. Key changes included: 1) retention of recurrent genetic abnormalities and the addition of new genetic mutations. Eight genes were included in the adverse risk category and designated as AML with myelodysplasia-related gene mutations. 2) The prognostic division based on the allelic ratio of FLT3-ITD in cases of AML coexisting with mutated NPM1 and FLT3-ITD was eliminated in ELN2017. This is due to the lack of standardization in the method for measuring the FLT-ITD allelic ratio. 3) Additionally, NPM1 mutated AML with additional adverse-risk cytogenetic abnormalities was classified as an adverse risk. 4) Mutations in the basic leucine zipper region of CEBPA that affect in-frame confer a favorable prognosis, regardless of whether monoallelic or biallelic mutations. 5) Additional cytogenetic abnormalities such as t(3q26.2;v)/MECOME-rearranged and t(8;16)(p11.2;p13.3)/KAT6A::CREBBP fusion, are now included in the adverse risk group [25, 26]. 6) Hyperdiploidy with multiple trisomies is not considered a complex karyotype.
Conclusion
In recent years, studies on the genetic spectrum of AML have increased, expanding the treatment possibilities [27]. Various gene-targeted therapies, such as FLT3 inhibitors, are being introduced in chemotherapy regimens and are undergoing continuous clinical trials [28, 29]. While both the WHO2022 and ICC classification systems are based on these findings, these new classifications have added complexity for researchers and physicians. Differences in terminology and the introduction of updated/new diagnostic entities can cause confusion in the field, affecting diagnosis, management, clinical outcome assessment, and clinical trials [2, 3]. Additionally, the diagnosis and risk stratification of AML require various molecular tests, which depend on the adequate economic and diagnostic capacity for their execution. Molecular tests, taking more than two weeks for a formal report, may result in a delayed diagnosis compared to traditional morphological diagnoses, potentially delaying treatment. The new classification system integrates morphological, immunophenotypic, molecular, and cytogenetic information, facilitating the adoption of precision medicine. Consequently, treatment decisions should be based on comprehensive laboratory tests, medical histories, and clinical information.
Availability of data and materials
No datasets were generated or analysed during the current study.
References
Appelbaum FR. WHO, what, when, where, and why: new classification systems for acute myeloid leukemia and their impact on clinical practice. Best Pract Res Clin Haematol. 2023;36:101518.
Falini B, Martelli MP. Comparison of the International Consensus and 5th WHO edition classifications of adult myelodysplastic syndromes and acute myeloid leukemia. Am J Hematol. 2023;98:481–92.
Shallis RM, Daver N, Altman JK, et al. Standardising acute myeloid leukaemia classification systems: a perspective from a panel of international experts. Lancet Haematol. 2023;10:e767–76.
Weinberg OK, Porwit A, Orazi A, et al. The International Consensus Classification of acute myeloid leukemia. Virchows Arch. 2023;482:27–37.
Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–8.
Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. 3rd ed. Lyon, France: IARC Press; 2001.
Swerdlow SH, Campo E, Harris NL, et al editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC Press; 2008.
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19.
Falini B, Brunetti L, Sportoletti P, Martelli MP. NPM1-mutated acute myeloid leukemia: from bench to bedside. Blood. 2020;136:1707–21.
Hwang SM. Classification of acute myeloid leukemia. Blood Res. 2020;55:S1–4.
Falini B, Martelli MP, Bolli N, et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011;117:1109–20.
Montalban-Bravo G, Kanagal-Shamanna R, Sasaki K, et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv. 2019;3:922–33.
Forghieri F, Nasillo V, Paolini A, et al. NPM1-mutated myeloid neoplasms with< 20% blasts: a really distinct clinico-pathologic entity? International J Mol Sci. 2020;21:8975.
Su L, Shi Y-Y, Liu Z-Y, Gao S-J. Acute myeloid leukemia with CEBPA mutations: current progress and future directions. Front Oncol. 2022;12:806137.
Snaddon J, Smith ML, Neat M, et al. Mutations of CEBPA in acute myeloid leukemia FAB types M1 and M2. Genes Chromosomes Cancer. 2003;37:72–8.
Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26:1549–56.
Bertrums EJ, Smith JL, Harmon L, et al. Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica. 2023;108:2044.
Gao Y, Jia M, Mao Y, et al. Distinct mutation landscapes between acute myeloid leukemia with myelodysplasia-related changes and de novo acute myeloid leukemia. Am J Clin Pathol. 2022;157:691–700.
Tazi Y, Arango-Ossa JE, Zhou Y, et al. Unified classification and risk-stratification in acute myeloid leukemia. Nat Commun. 2022;13:4622.
Alexander TB, Orgel E. Mixed phenotype acute leukemia: current approaches to diagnosis and treatment. Curr Oncol Rep. 2021;23:1–10.
Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–77.
Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Kayser S, Hills RK, Langova R, et al. Characteristics and outcome of patients with acute myeloid leukaemia and t (8; 16)(p11; p13): results from an International Collaborative Study. Br J Haematol. 2021;192:832–42.
Ottema S, Mulet-Lazaro R, Beverloo HB, et al. Atypical 3q26/MECOM rearrangements genocopy inv (3)/t (3; 3) in acute myeloid leukemia. Blood. 2020;136:224–34.
Short NJ, Tallman MS, Pollyea DA, Ravandi F, Kantarjian H. Optimizing risk stratification in acute myeloid leukemia: dynamic models for a dynamic therapeutic landscape. J Clin Oncol. 2021;39:2535.
Ahn J-S, Kim H-J. FLT3 mutations in acute myeloid leukemia: a review focusing on clinically applicable drugs. Blood Res. 2022;57(S1):32–3.
Kim D-S, Byun J-M, Shin D-Y, et al. Concomitant ruxolitinib with cytarabine-bassed induction chemotherapy in secondary acute meyloid leukemia evolving from myeloproliferative neoplasm. Blood Res. 2023;58(3):155–7.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
H.S.Park wrote the main manuscript text.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This review did not require approval from the relevant institutional review board or ethics committee.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, H.S. What is new in acute myeloid leukemia classification?. Blood Res. 59, 15 (2024). https://doi.org/10.1007/s44313-024-00016-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44313-024-00016-8