
Abstract
Badnaviruses are dsDNA plant viruses that cause diseases in economically important crops in most tropical and subtropical regions of the world. In northeastern Brazil, badnaviruses are a serious constraint to yam crops. We assessed the genetic variability of badnaviruses infecting yam crops in northeastern Brazil, based on the sequence of the RT/RNAseH domain of 96 badnavirus isolates obtained between 2011 and 2012. Phylogenetic analysis revealed that Dioscorea bacilliform AL virus (DBALV) is the only badnavirus species infecting yam in the Brazilian northeast part. No evidence of recombination was found among DBALV isolates. Genetic variability of the DBALV population is high, with a mutation frequency on the order of 10−4, similar to those found for RNA viruses. We conclude that several mechanisms, including purifying selection and adaptation of the virus to the dioscorea species cultivation system, are important evolutionary processes in the genetic diversification of the DBALV population.
Similar content being viewed by others


Introduction
The family Caulimoviridae comprises plant viruses with a double-stranded (ds) DNA genome that replicate through an RNA intermediate (Geering and Hull 2012). Based on their genome organization, host range, insect vector and nucleotide sequence identity, members of Caulimoviridae are classified into seven genera: Badnavirus, Caulimovirus, Cavemovirus, Petuvirus, Solendovirus, Soymovirus and Tungrovirus (Geering and Hull 2012). Viruses belonging to the genus Badnavirus have a genome comprised of a single circular dsDNA of about 7.0–7.6 kbp encapsidated in a non-enveloped bacilliform particle. Each DNA strand has discontinuities at specific sites. The genomes of all badnaviruses possess three open reading frames (ORFs) giving origin to three proteins, P1, P2 and P3. The function of P1 is unknown; P2 is the virion-associated capsid protein. Polyprotein P3 contains several functional domains: cystein rich (CYS), RNA-binding (RB), aspartate protease (PR) and viral replicase [reverse transcriptase (RT) and ribonuclease H (RNaseH)] (Medberry et al. 1990; Harper and Hull 1998; Geering and Hull 2012). The demarcation of badnavirus species is based on host range, vector specificities and the determination of the nucleotide sequence of the RT/RNaseH genomic region. The International Committee on Taxonomy of Viruses (ICTV) has established a species demarcation threshold of ≥80 % nucleotide identity for the RT/RNaseH (Geering and Hull 2012).
Badnaviruses are transmitted in a semi-persistent manner by mealybugs (Geering and Hull 2012) to a wide range of economically important tropical crops, including yams (Dioscorea spp.) (Phillips et al. 1999). The first report of badnaviruses in yam occurred in Barbados in association with brown staining in tubers of Dioscorea alata L. (a disease that requires also the presence of a member of the genus Potyvirus) (Harrison and Roberts 1973). Yam-infecting badnaviruses are widespread in tropical regions, particularly in Africa and in the South Pacific (Seal and Muller 2007; Eni et al. 2008; Kenyon et al. 2008). Currently, two badnavirus species that infect yam are recognized by the ICTV: Dioscorea bacilliform AL virus (DBALV) and Dioscorea bacilliform SN virus (DBSNV), obtained from D. alata and D. sansibarensis Pax, respectively (Geering and Hull 2012). However, studies carried out in Africa and the South Pacific based on RT/RNaseH sequences revealed a high incidence and diversity of yam-infecting badnaviruses (Eni et al. 2008; Kenyon et al. 2008). This fact has been attributed to the many different species of Dioscorea found in these areas, to their vegetative mode of propagation, which contributes to virus accumulation, and to the unrestricted exchange of plant material (Eni et al. 2008; Kenyon et al. 2008; Bousalem et al. 2009).
Brazil is the second major producer of yam in the Americas after Colombia, with a production of 244,142 t in 2012 (FAO 2010). Badnaviruses are limiting factors to production, and have been described infecting the two most important commercial cultivated species of yams: São Tomé (D. alata) and da Costa (D. cayennensis Lam.). In contrast with what is found in Africa and Southern Asia, DBALV seems to be the only badnavirus infecting yam, being widely distributed in the northeastern region (Lima et al. 2013).
Determining the genetic variability of a viral population is important to understand how these populations evolve, as well as the implications for durability of control strategies (Seal et al. 2006). Evidence has accumulated indicating that RNA viruses and retroviruses evolve quickly due to the high mutation rates and lack of proofreading activity of their RNA-dependent RNA polymerases and reverse transcriptases (Drake 1993; Drake and Holland 1999; Bebenek et al. 1999). Since dsDNA viruses encode less error-prone polymerases or rely on proofreading host-encoded DNA polymerases and repair systems, they are generally less variable in their sequences (Duffy et al. 2008; Garcia-Diaz and Bebenek 2007; Kunkel and Erie 2005; McCulloch and Kunkel 2008).
According to Firth et al. (2010), dsDNA viruses are frequently described to evolve through long-term co-divergent associations with their hosts, where a pattern of low rates of nucleotide substitution is expected. Rates of molecular evolution estimated for Herpes simplex virus (HSV; 3.5 × 10−8) and Human papilloma virus (HPV; 4.5 × 107) are low, reinforcing the hypothesis of co-divergence with human populations (Sakaoka et al. 1994; Ong et al. 1993). However, a high mutation rate of approximately 1.0 × 10−5 substitutions/site/year was observed for Variola virus (VARV) (Firth et al. 2010). Rates between 1.71 and 5.81 × 10−4 substitutions/site/year were observed for Cauliflower mosaic virus (CaMV, the type species of the genus Caulimovirus), approaching that of many RNA and ssDNA viruses (Yasaka et al. 2014). Little is known about the variability within dsDNA plant viruses, especially badnaviruses. Considering that these viruses replicate using reverse transcription, it is possible that their genetic variability is closer to the one of retroviruses than to dsDNA viruses which replicate using DNA-dependent DNA polymerases.
The purpose of this study was to assess the genetic variability of yam badnaviruses in northeastern Brazil.
Material and methods
Sample collection and storage
Foliar samples with virus-like symptoms such as mosaic, leaf distortion, shoestring and stunting were collected in five yam fields throughout the states of Alagoas (AL), Paraíba (PB) and Pernambuco (PE) in 2011–2012 (Table 1). Leaves were placed in paper bags and stored at −80 °C.
DNA extraction and viral genome amplification
Total DNA extraction was carried out from fresh or frozen (−80 °C) leaves according to Doyle and Doyle (1987). The DNAs were used as templates for PCR amplification using the degenerate primer set Badna-FP (5′- ATG CCI TTY GGI ITI AAR AAY GCI CC-3′) and Badna-RP (5′-CCA YTT RCA IAC ISC ICC CCA ICC-3′), designed based on the RT/RNaseH coding sequences of members of the genus Badnavirus (Yang et al. 2003).
PCR reactions were performed in a volume of 60 μL containing 6 μL of 10X buffer (100 mM KCl, 100 mM Tris–HCl pH 9.0, 1 % Triton-X100), 4.8 μL of dNTPs (2.5 mM), 1.8 μL of MgCl2 (50 mM), 3 μL of each primer (10 μM), 1U Taq DNA polymerase (Life Technologies), 1 μL (10 to 100 ng) of template DNA and 40.2 μL ultrapure water. PCR cycling was performed in an Applied Biosystems 2720 thermal cycler programmed as follows: initial denaturation at 94 °C for 4 min, followed by 35 cycles at 94 °C for 30 s, 50 °C for 30 s and 72 °C for 1 min, and one final cycle at 72 °C for 10 min. Amplicons of the expected size (579 bp) were purified from agarose gels using the GFX PCR DNA and Gel Band Purification kit (GE Healthcare) according to the manufacturer’s instructions. Sequencing was performed by Macrogen Inc. (Seoul, South Korea) directly from the gel-purified PCR products using forward and reverse primers.
Sequence comparisons and phylogenetic analysis
Sequences of the RT/RNaseH region were initially analyzed with the BLASTn algorithm (Altschul et al. 1990) to determine the viral species with which they shared the greatest similarity.
To determine the taxonomic placement of viral isolates, additional pairwise nucleotide sequence comparisons between sequences obtained in this study, other species of badnavirus available in GenBank and a DBALV sequence dataset comprising isolates obtained from D. alata and D. cayenensis plants collected in the states of AL, PB and PE in 2011 (Table 2; Lima et al. 2013) were performed with the program Species Demarcation Tool (SDT) v. 1.0 (Muhire et al. 2013).
Multiple nucleotide sequence alignments of partial nucleotide sequences of the RT/RNaseH regions were prepared using MUSCLE (Edgar 2004). Phylogenies were reconstructed using maximum-likelihood (ML) with RAxML v.7.0.3 (bioinformatics.oxfordjournals.org/content/22/21/2688.long) and the General Time Reversible (GTR) nucleotide substitution model with gamma distribution rate of heterogeneity. The RT/RNaseH sequences were chosen for analysis because of their essential role for viral replication, therefore being subject to stricter variability constraints (Geering and Hull 2012). However, no conclusion drawn for this region can be extended to other genomic regions. Robustness of each internal branch was estimated by bootstrapping (1000 replications). Trees were visualized using the Tree View program (Page 1996) and edited using CorelDraw X3.
Recombination analysis
Evidence of non-tree-like evolution was assessed for RT/RNaseH dataset using the Neighbor-Net method implemented in the program SplitsTree4 (Huson and Bryant 2006). Analysis of potential recombination events was carried out using the recombination detection methods rdp, Geneconv, Bootscan, Maximum Chi Square, Chimaera and Sister Scan implemented in the Recombination Detection Program (RDP) ver. 3.0 (Martin et al. 2010) using default parameters, except that sequences were considered linear.
General descriptors of the genetic structure of viral populations
The RT/RNaseH dataset was used to assess the partition of genetic variability and population structure based on Wright’s F fixation index (Weir 1996).
The main descriptors of molecular variability were estimated, including the total number of segregating sites (s), total number of mutations (Eta), average number of nucleotide differences between sequences (k), nucleotide diversity (π), mutation frequencies, number of haplotypes (h) and haplotype diversity (Hd). These analyses were performed using the program DnaSP v. 5 (Rozas et al. 2003).
Parameterization of evolutionary mechanisms
Four types of neutrality tests were used to verify the occurrence of selection in populations: Tajima’s D, Fu and Li’s D* and F* and the test based on the number of synonymous (dS) and non-synonymous (dN) substitutions with the Pamilo-Bianchi-Li (PBL) model. These analyses were performed using the program DnaSP v. 5, with different sets of data considering unique populations or subpopulations separated on the basis of the year of collection, geographical location and host.
To detect amino acid sites under positive and negative selection, amino acid sequence data sets were analyzed using the single-likelihood ancestor counting (SLAC) method implemented in the DataMonkey server (www.datamonkey.org).
Results
Sequence data set
A total of 150 samples were collected, of which 108 were positive for the presence of a badnavirus based on the detection of a PCR fragment of 579 bp corresponding to the RT/RNaseH domains, located in the C-terminal part of the ORF3. All positive samples were sequenced, and contigs of good quality and length of 435 pb were assembled for 48 sequences.
Sequence comparisons and phylogenetic analysis
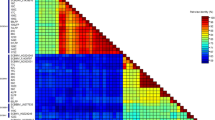
Using pairwise comparisons of the RT/RNaseH sequences and the ≥80 % nucleotide identity criterion established by the ICTV (Geering and Hull 2012), the 48 isolates described here were assigned to only one badnavirus species: DBALV (Fig. 1). These results were confirmed by phylogenetic analysis, in which all isolates from this study and the 48 isolates from Lima et al. (2013) clustered in a single monophyletic branch with isolates of DBALV, supported by a bootstrap value of 90 % (Fig. 2). These sequences were considered as representative sequences of the isolates present in the samples, and therefore collectively referred as populations.

Two-dimensional graphic representing the percentage of pairwise sequence identity for the nucleotide sequences of the RT/RNaseH between the badnavirus isolates obtained in this work and the isolates reported by Lima et al. (2013) and other badnavirus species with sequences available in GenBank

Maximum likelihood phylogenetic tree based on the sequence of the RT/RNaseH region of DBALV isolates obtained in this work (indicated in blue), from Lima et al. (2013) (red) and other badnavirus species with sequences available in GenBank (black). Rice tungro bacilliform virus (RTBV, genus Tungrovirus) was used as the outgroup
Recombination analysis
Occurrence of recombination events among DBALV isolates and other badnavirus species was initially tested by Neighbor-Net analysis. The results did not indicate any significant evidence of intra- or interspecies recombination (data not shown).
To further investigate recombination within these sequences, a data set including badnavirus species available in GenBank, the 48 sequences from this study plus the 48 sequences from Lima et al. (2013) was analyzed using the RDP3 package. However no evidence of recombination was observed, confirming the Neighbor-Net result.
General descriptors of the genetic structure of viral populations
The DBALV isolates were initially divided into putative subpopulations according to the year of collection (2011 or 2012), geographic region (Alhandra, Arapiraca, Bonito, Chã Preta or Viçosa) and host species (D. alata or D. cayenensis). The value obtained for the Fst test was statistically significant only for the year of collection, indicating that these two subpopulations were actually structured.
The DBALV population obtained in this work has a high degree of genetic variability, with a nucleotide diversity (π) for the RT/RNAseH region of 0.05342 and a mutation frequency on the order of 10−4. The 2011 subpopulation is more diverse than the 2012 subpopulation, with higher values for every descriptor (Table 3).
Parameterization of evolutionary mechanisms
Neutrality tests were used to assess what kind of selection or demographic forces are acting upon the DBALV population. The values obtained for Tajima’s D, Fu and Li’s D* and F* tests were not statistically significant (Table 4). The SLAC method detected dN/dS values <1, with 58 sites found to be evolving under negative selection versus 0 sites evolving under positive selection. These results are indicative of purifying selection acting on this population.
Discussion
Diseases caused by viruses of the genus Badnavirus are responsible for major damage to yam crops in many tropical regions, including Brazil (Seal and Muller 2007; Eni et al. 2008; Kenyon et al. 2008; Bousalem et al. 2009; Lima et al. 2013). A recent study about badnaviruses infecting yam in northeastern Brazil suggested that DBALV may be the only species occurring in this region, and that its sequence variability is high (Lima et al. 2013). Here, we have studied the genetic structure and variability of yam badnavirus populations in detail. Our results showed that DBALV was the only yam-infecting badnavirus, corroborating the data obtained by Lima et al. (2013) and reinforcing that DBALV seems to be the only badnavirus infecting this crop in northeastern Brazil. Factors such as constant exchange of infected plant material between areas can be contributing to the predominance of DBALV in this region. Conversely, studies performed in Africa and the South Pacific suggest the presence of up to twelve species of badnaviruses in yam (Eni et al. 2008; Kenyon et al. 2008; Bousalem et al. 2009).
Recombination events can result in changes in the biological properties of a virus, such as the expansion of its host range (Gibbs and Weiller 1999), virulence (Zhou et al. 1997; Pita et al. 2001) and fitness (Monci et al. 2002). Recombination among members of the Caulimoviridae family is of particular interest, as these viruses are able to integrate into the host plant genome as endogenous pararetroviral sequences (EPRVs) (Squires et al. 2011). In some cases, EPRVs can be activated by homologous recombination resulting in the episomal form of the virus which causes systemic infection of the host. Homologous recombination events have been reported for the activation of the episomal genome of CaMV, Banana streak virus (BSV) and Tobacco vein clearing virus (TVCV) (Gal et al. 1991; Ndowora et al. 1999; Froissart et al. 2005; Staginnus and Richert-Poggeler 2006). The recombination rate of a CaMV population was experimentally estimated to be on the order of 10−5, indicating that it is a very frequent event for this virus (Froissart et al. 2005). Nevertheless, we did not find evidence of recombination among the DBALV isolates. A recombination event was detected between two yam-infecting badnaviruses, Dioscorea esculenta bacillifom virus A (DeBV-A) and Dioscorea esculenta bacilliform virus B (DeBV-B), and was located in the RT region (Bousalem et al. 2009). The relatively short length of the fragment analyzed in our study (435 bp, in a genome with over 7.0 kbp) could be one of the reasons for the failure to detect any recombination event.
Analysis of the DBALV population demonstrated that its genetic variability is indeed very high (Table 3), to a level which is similar to that for RNA viruses. It was also slightly higher than that found for a population of genotype F of Hepatitis B virus (a human dsDNA, reverse-transcribing virus in the family Hepadnaviridae) obtained in the northern region in Brazil (Mello et al. 2013; Table 3). Interestingly, the variability of the DBALV 2011 subpopulation was higher than that of the 2012 subpopulation. This could reflect local variations in cultivated and non-cultivated host distribution at sampling sites or the presence of distinct vector populations.
Mutation and substitution rates commonly observed for retroviruses are considerably higher than those observed for non-reverse transcribing dsDNA viruses, and most likely reflect differences between the fidelity of reverse transcriptases and DNA-dependent DNA polymerases (Ong et al. 1993; Sakaoka et al. 1994; McGeoch et al. 2000; Zhou and Holmes 2007; Mello et al. 2013). Recently, substitution rates between 1.71 and 5.81 × 10−4 subs/site/year were observed for CaMV (Yasaka et al. 2014). We estimated a mutation frequency for the DBALV population similar to the value estimated for the hepadnavirus HBV (Mello et al. 2013). Although the mutation frequencies calculated for DBALV and HBV cannot be directly compared with the substitution rates determined by Yasaka et al. (2014), it is noteworthy that the mutation frequency determined for DBALV is very high. Although there is no precise measure of the fidelity of the hepadnavirus RT, reverse transcriptases are known to be highly error prone in all retroviruses and other retroelements for which a rate has been estimated (Drake et al. 1998; Svarovskaia et al. 2003).
Neutrality tests and dN/dS ratio indicated that the DBALV population may be under purifying selection, or that a recent expansion is operating at both the nucleotide and protein levels. Purifying selection acting upon the RT/RNaseH could emphasize the critical function performed by this protein and its requirement for DBALV survival. Purifying selection was also the major evolutionary force acting on the reverse transcriptase domain of HBV in humans (Osiowy et al. 2006). However, the occurrence of mutations is not sufficient to fully explain the genetic variability of the DBALV population, reinforcing a possible influence of other evolutionary forces such as migration and recombination.
We conclude that the high genetic variability observed for DBALV populations may reflect its ability to evolve rapidly under the yam cultivation system implemented in Brazil. This should be taken into account in cases of implementation of control measures based on genetic resistance, as well as in the use of detection methods based on genome sequence, since both could fail due to the rapid evolution of this virus.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bebenek K, Boyer JC, Kunkel TA (1999) The base substitution fidelity of HIV-1 reverse transcriptase on DNA and RNA templates probed with 8-oxo-deoxyguanosine triphosphate. Mutat Res 429:149–158
Bousalem M, Durand O, Scarcelli N, Lebas BSM, Kenyon L, Marchand JL, Lefort F, Seal SE (2009) Dilemmas caused by endogenous pararetroviruses regarding the taxonomy and diagnosis of yam (Dioscorea spp.) badnaviruses: analyses to support safe germplasm movement. Arch Virol 154:297–314
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull 19:11–15
Drake JW (1993) Rates of spontaneous mutations among RNA viruses. Proc Natl Acad Sci U S A 90:4171–4175
Drake JW, Holland JJ (1999) Mutation rates among RNA viruses. Proc Natl Acad Sci U S A 96:13910–13913
Drake JW, Charlesworth B, Charlesworth D, Crow JF (1998) Rates of spontaneous mutation. Genetics 148:1667–1686
Duffy S, Shackelton LA, Holmes EC (2008) Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet 9:267–276
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf 5:1–19
Eni AO, Hughes J’A, Rey M (2008) Sequence diversity among badnavirus isolates infecting yam (Dioscorea spp.) in Ghana, Togo, Benin and Nigeria. Arch Virol 153:2263–2272
FAO (2010) FAOSTAT - agricultural statistics database. Available at: <http://faostat.fao.org/default.aspx>. Accessed on December 14, 2013
Firth C, Kitchen A, Shapiro B, Suchard MA, Holmes EC, Rambaut A (2010) Using time-strutured data to estimate evolutionary rates of double-stranded DNA viruses. Mol Biol Evol 27:2038–2051
Froissart R, Roze D, Uzes M, Galibert L, Blanc S, Michalakis Y (2005) Recombination every day: abundant recombination in a virus during a single multi-cellular host infection. PLoS Biol 3:389–395
Gal S, Pisan B, Hohn T, Grimsley N, Hohn B (1991) Genomic homologous recombination in planta. EMBO J 10:1571–1578
Garcia-Diaz M, Bebenek K (2007) Multiple functions of DNA polymerases. Crit Rev Plant Sci 26:105–122
Geering ADW, Hull R (2012) Family Caulimoviridae. In: King AMQ, Adams MJ, Cartens EB, Lefkowitz EJ (eds) Virus taxonomy. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, pp 429–443
Gibbs MJ, Weiller GF (1999) Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc Natl Acad Sci U S A 96:8022–8027
Harper G, Hull R (1998) Cloning and sequence analysis of Banana streak virus DNA. Virus Genes 17:271–278
Harrison BD, Roberts IM (1973) Association of virus-like particles with internal brown spot of yam (Dioscorea alata). Trop Agric 50:335–340
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Kenyon L, Lebas BSM, Seal SE (2008) Yams (Dioscorea spp.) from the South Pacific Islands contain many novel badnaviruses: implications for international movement of yam germplasm. Arch Virol 153:877–889
Kunkel TA, Erie DA (2005) DNA mismatch repair. Annu Rev Biochem 74:681–710
Lima JS, Lima ATM, Castillo-Urquiza GP, Silva SJC, Assunção IP, Michereff SJ, Zerbini FM, Lima GSA (2013) Variabilidade genética de isolados de badnavírus infectando inhame (Dioscorea spp.) no nordeste do Brasil. Trop Plant Pathol 38:349–353
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
McCulloch SD, Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18:148–161
McGeoch DJ, Dolan A, Ralph AC (2000) Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol 74:10401–10406
Medberry SL, Lockhart BEL, Olszewski (1990) Properties of Commelina yellow mottle virus complete DNA sequence, genomic discontinuities and transcript suggest that it is a pararetrovirus. Nucleic Acids Res 18:5505–5513
Mello FCA, Araújo OC, Lago BV, Motta-Castro ARC, Moraes MTB, Gomes SA, Bello G, Araújo NM (2013) Phylogeography and evolutionary history of hepatitis B virus genotype F in Brazil. Virol J 10:236–243
Monci F, Sánchez-Campos S, Navas-Castillo J, Moriones E (2002) A natural recombinant between the geminiviruses Tomato yellow leaf curl Sardinia virus and Tomato yellow leaf curl virus exhibits a novel pathogenic phenotype and is becoming prevalent in Spanish populations. Virology 303:317–326
Muhire B, Martin DP, Brown JK, Navas-Castillo J, Moriones E, Zerbini FM, Rivera-Bustamante R, Malathi VG, Briddon RW, Varsani A (2013) A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch Virol 158:1411–1424
Ndowora T, Dahal G, LaFleur D, Happer G, Hull R, Olszewski NE, Lockhart BEL (1999) Evidence that badnavirus infection in Musa can originate from integrated pararetroviral sequences. Virology 255:214–220
Ong CK, Chan SY, Campo MS, Fujinaga K, Mayromara-Nazos P, Labropoulou V, Pfister H, Tay SK, Meulen J, Villa LL (1993) Evolution of human papillomavirus type 18: an ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J Virol 67:6424–6431
Osiowy C, Giles E, Tanaka Y, Mizokami M, Minuk GY (2006) Molecular evolution of hepatitis B virus over 25 years. J Virol 80:10307–10314
Page RD (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Phillips S, Briddon RW, Brunt A, Hull R (1999) The partial characterization of a badnavirus infecting the greater Asiatic or water yam (Dioscorea alata). J Phytopathol 147:265–269
Pita JS, Fondong VN, Sangare A, Otim-Nape GW, Ogwal S, Fauquet CM (2001) Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J Gen Virol 82:655–665
Rozas J, Sánchez-Delbarrio JC, Messeguer X, Rozas R (2003) DNAsp: DNA polymorphism analyses by coalescent and other methods. Bioinformatics 19:2496–2497
Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim YT, Ren CS, Nahmias AJ (1994) Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J Gen Virol 75:513–527
Seal S, Muller E (2007) Molecular analysis of a full-length sequence of a new yam badnavirus from Dioscorea sansibarensis. Arch Virol 7:1–7
Seal SE, Van den Bosch F, Jeger MJ (2006) Factors influencing begomovirus evolution and their increasing global significance: implications for sustainable control. Crit Rev Plant Sci 25:23–46
Squires J, Gillespie T, Schoelz JE, Palukaitis P (2011) Excision and episomal replication of cauliflower mosaic virus integrated into a plant genome. Plant Physiol 155:1908–1919
Staginnus C, Richert-Poggeler KR (2006) Endogenous pararetroviruses: two faced travelers in the plant genome. Trends Plant Sci 11:485–491
Svarovskaia ES, Cheslock SR, Zhang WH, Hu WS, Pathak V (2003) Retroviral mutation rates and reverse transcriptase fidelity. Front Biosci 8:d117–d134
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer Associates Inc., Sunderland
Yang IC, Hafner GJ, Revill PA, Dale JL, Harding RM (2003) Sequence diversity of South Pacific isolates of Taro bacilliform virus and the development of a PCR-based diagnostic test. Arch Virol 148:1957–1968
Yasaka R, Nguyen HD, Ho SYW, Duchene S, Korkmaz S, Katis N, Takahashi H, Gibbs AJ, Ohshima K (2014) The temporal evolution and global spread of Cauliflower mosaic virus, a plant pararetrovirus. PLoS ONE 9:1–12
Zhou Y, Holmes E (2007) Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J Mol Evol 65:197–205
Zhou X, Liu Y, Calvert L, Munoz C, Otim-Nape GW, Robinson DJ, Harrison BD (1997) Evidence that DNA-A of a geminivirus associated with severe cassava mosaic disease in Uganda has arisen by interspecific recombination. J Gen Virol 78:2101–2111
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Alice Nagata
Rights and permissions
About this article

Cite this article
Guimarães, K.M.C., Silva, S.J.C., Melo, A.M. et al. Genetic variability of badnaviruses infecting yam (Dioscorea spp.) in northeastern Brazil. Trop. plant pathol. 40, 111–118 (2015). https://doi.org/10.1007/s40858-015-0023-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40858-015-0023-y