
Abstract
Decline in regenerative capacity due to depletion or dysfunction of stem cells is a prominent hallmark of aging across tissues and organisms. Molecular damages such as protein, organelle, and DNA damages are commonly recognized as drivers of age-related stem cell decline. In this review, we discuss diverse damage types and respective avoidance strategies used by stem cells across species with specific focus on avoidance of genotoxic damage and evasion of DNA-damage induced death. Within DNA damage management by stem cells, protective strategies range from asymmetric damage exclusion to downregulation of DNA damage checkpoints and upregulation of pro-survival factors. We also discuss systemic impact of stem cell DNA damage from the angle of its organ-damaging role and from the unexpected pro-homeostatic angle. We use examples of progeroid syndromes and acute irradiation exposure to outline the impact of stem cell DNA damage on the aging process.
Similar content being viewed by others

Introduction: Adult Stem Cells as Hubs of Damage Accumulation During Aging
Aging is commonly regarded as a process of longitudinal functional decline leading to eventual complete deterioration of vital body functions. Precise underlying mechanisms of the deterioration process are still highly controversial. However, it is well documented that during the lifetime, tissues are targeted by numerous molecular damages originating from the outer environment as well as from inside the cells. Typical examples of intracellular damages are replication-driven DNA damage, reactive oxygen species (ROS) produced by dysfunctional mitochondria, and harmful products of de-regulated metabolism [1]. With age, the stringency of metabolic regulation progressively declines as does mitochondrial integrity and replication fidelity [2, 3]. Thus, the level of internal molecular damages gradually grows. At the same time, the efficiency of intracellular quality control and repair machineries declines leaving cells with progressively increasing damage loads [4]. The exposure to unbearable amount of molecular damages leads to age-related death of cells across tissues rendering tissues highly dependent on the replenishing process orchestrated by respective adult stem cells and making stem cell self-renewal a critical factor for unperturbed organ homeostasis during aging. While differentiated cells turnover with high frequencies during lifetime, stem cells reside in their respective niches for the whole organismal life. This means adult stem cells are exposed to internal and external damages for the longest time among all cells. Yet, considering their key role in organ regeneration, stem cells are expected to withstand higher damage loads compared to somatic cells. How stem cells cope with increasing damage levels during aging and how particular niches regulate this process will be the topic of the current review. We will also discuss how damage-driven stem cell dysfunction impacts on organ homeostasis during aging.
Coping with Age-Related Damage of Proteins and Organelles—The Asymmetric Distribution
Large fraction of molecular damages experienced by stem cells with time is protein damages [5] leading to protein misfolding and formation of toxic protein aggregates. Damages of membrane organelles including mitochondria are another prominent kind [6]. In budding yeast, a classical model of replicative aging, it was observed that damaged organelles and proteins are not distributed equally during cell division: while one resulting cell regarded as “mother” cell retains all damage, the second “daughter” cell remains damage free. Subsequently, “mother” cells demonstrate gradual decline of cellular functions and “daughter” cells remain functionally young. Asymmetric segregation during yeast mitosis has been described for oxidized proteins, protein aggregates, and faulty mitochondria [7–9]. It is indeed an active process depending on actin cytoskeleton, chaperon abundance, and activity of anti-aging molecules such as conserved NAD-dependent histone deacetylase Sirt2 [10]. Few years later, asymmetric partitioning of damaged proteins during stem cell division was uncovered in Drosophila. The phenomenon was observed in the germline and in the intestine of adult flies [11, 12]. It was found that germline stem cells retain damage similar to yeast “mother” cells while intestinal stem cells re-distribute altered molecules to differentiating enteroblasts. In both tissues, signals from respective niches (linked to elements of extracellular matrix) were thought to determine damage retention or exclusion. The intriguing difference in damage handling between germline and intestinal stem cells is thought to originate from their distinct physiological functions. Since germline gives rise to new organisms expected to outlive the parent, germline “daughters” require higher fitness compared to “mothers”. On the other hand, differentiated intestinal cells are short lived and rapidly replenished by the stem cell pool which in turn needs to remain fit to last throughout the life of an animal. Thus, longevity of germline stem cells is inferior to the fitness of the progeny (conceptually similar to fate of “mother” cells in yeast), while longevity of intestinal stem cells (ISCs) is critical for the survival of the fly. Such distinct fitness requirements determine differential damage retention strategies between the two stem cell types which are presumably regulated by their respective niche signals.
Recently, asymmetric partitioning of damaged proteins and mitochondria was demonstrated also in mammalian stem cells. By following fates of differentially labeled “young” and “old” organelles during division of human mammary epithelium stem-like cells (SLCs), researchers observed asymmetric accumulation of “old” membrane material in one of two daughter cells. High levels of old membranes (mostly mitochondria) coincided with differentiated phenotype of the daughters while damage free daughters maintained stem cell traits [13•]. Asymmetric distribution of young and old mitochondria strongly depended on intact mitochondrial fission and fusion machinery of SLCs. These data suggest that mammary epithelium, a tissue with high need of stem-cell driven remodeling, behaves comparable to the intestine of adult flies in maintaining fitness of stem cells at a cost of increased damage to differentiated daughters. Evidence of asymmetric damage distribution was also obtained by in vitro studies of murine neural stem cells (NSCs). It was found that during division of embryonic and young adult NSCs altered proteins are inherited predominantly by the daughter that is destined to differentiate [14•]. This process is orchestrated by a lateral diffusion barrier in the membrane of the endoplasmic reticulum which pre-segregates cellular components prior to cell division. With age, the barrier function appears to decline due to nuclear lamin deterioration leading to loss of asymmetric damage distribution that may be one of the driving forces of brain aging. Different damage evasion strategies of distinct stem cell types are summarized in Table 1.
Coping with Age-Related DNA Damage—Does Asymmetric Partitioning Work for DNA?
In addition to protein and organelle damages, another critical type of damage comes from the very core of stem cell self-renewal function—the replication of DNA. Faulty DNA replication is believed to be the key source of genotoxic damage during aging [17]. Experiencing more cell divisions per cellular lifetime than the majority of other body cells, stem cells are expected to have a strategy to fight DNA damage accumulation. Could this strategy rely on asymmetric partitioning of genotoxic damage similar to damaged organelles and proteins? This has been an intriguing question for a long time. The existence of asymmetric partitioning was clearly demonstrated for damaged mitochondrial DNA: In yeast, mitochondria with damaged genomes were retained by mother cells along with other damages [8]. Faulty intra-chromosomal recombination often leads to generation of extra-chromosomal DNA circles whose strong contribution to cellular dysfunction during aging was demonstrated in yeast. It has been found that these aberrant DNA species are retained by mother cells. During replication process DNA circles remain attached to old nuclear pores that are kept in mother cells by a septin-dependent lateral diffusion barrier formed at the nuclear envelope [18]. The option of asymmetric distribution of damaged nuclear DNA has been debated for many years. In the 1960s, a phenomenon of non-random co-segregation of template DNA strands was described by using mouse and hamster cells [19]. This observation suggests that during asymmetric cell division, one daughter cell receives complete parental DNA while another daughter receives complete newly produced DNA (Table 1). This concept was immediately attractive with regard to stem cell function as it would indicate stem cells could reduce levels of DNA damage by excluding newly synthesized DNA molecules which have a higher chance of being altered. It was later suggested that also telomere length could be maintained by non-random DNA co-segregation process as telomere shortening occurs on newly produced chromatids. Asymmetric retention of parental DNA by stem cell-like daughters was indeed demonstrated for adult stem cells of skeletal muscle, skin, and intestine [20–23]. Template DNA and newly synthesized DNA receive distinct epigenetic marks [24]. It has been demonstrated that the particular epigenetic signatures have differential affinity to the nuclear envelope—previously outlined as an important player in asymmetric damage segregation [25]. It is thus possible that template DNA co-segregation during stem cell mitosis is driven by replication-linked epigenetic marks. In addition, to potential function in DNA segregation, epigenetic marks are likely to play an active role in cell fate decisions of daughter cells by inhibiting expression of self-renewal genes and by inducing differentiation. Intriguingly, the choice between symmetric self-renewal and asymmetric division often involves one of key cellular DNA damage sensors—the p53 molecule: high levels of p53 activity appear to shift stem cells to asymmetric divisions while p53 inhibition supports symmetric self-renewal [26]. These data suggest that p53 may act upon DNA damage to drive stem cells into protective damage segregation mode. Indeed, high p53 activity is associated with increased co-segregation of template DNA in some cellular systems [27]. Additionally, it has been found that DNA damage in hematopoietic and mammary stem cells leads to reduction of p53 activity below basal levels keeping cells in a self-renewal state [28•]. Lowering p53 activity may be important for rapid replenishing of damaged tissues which depends on fast expansion of the stem cell pool by self-renewal. It is thus possible that the choice between asymmetric and symmetric division upon genotoxic insult depends on the exact level of damage experienced by the tissue: in case of strong damage, stem cell self-renewal is favored while limited damages stimulate asymmetric damage exclusion. Importantly, asymmetric cell divisions and non-random DNA segregation appear to be regulated by patterns of extracellular matrix components produced by the niche [29•]. This is comparable to the regulatory background of asymmetric segregation of protein damage described in Drosophila. It is important to remember that non-random template DNA co-segregation in stem cells remains a highly controversial phenomenon despite being uncovered many years ago. The controversy is driven by incomplete reproducibility of data among comparable stem cell types which may arise from insufficiently uniform cell isolation methods. However, putative non-random DNA segregation shares conceptual and mechanistic features with well-known asymmetric processes in stem cells and thus remains a highly interesting hypothesis to be tested further. Moreover, stem cell DNA damage may influence asymmetric retention or exclusion of damaged organelles and proteins: high genotoxic burden leads to strong engagement of Poly (ADP-ribose) polymerase (PARP), a critical factor of DNA repair [30]. The activity of PARP critically depends on NAD+ consumption and leads to depletion of cellular NAD+ storages. This means other proteins depending on NAD+ cannot function in their full capacity. One of such proteins is Sirt2, a master regulator of protein damage proportioning found in yeast. It is thus feasible that high DNA damage loads negatively impact on all asymmetric processes in stem cells not only via p53 activity but also via NAD+ depletion [31].
Coping with Age-Related DNA Damage—Do DNA Damage Responses Work Differently in Stem Cells?
In general, cells with accumulated unrepaired DNA lesions are expected to be cleared by apoptotic death. Cell fate decision depends on activity of conserved cell-intrinsic checkpoints activated by DNA damage which drive irreversibly damaged cells into apoptosis or permanent cell cycle arrest through senescence [32]. However, in contrast to short-lived differentiated cells, adult stem cells are needed throughout organismal life to ensure tissue maintenance, a process critically dependent on the unique self-renewal capacity of stem cells. Considering this demand of functionality over longer periods of time, it is reasonable to expect a distinct, perhaps less stringent link between DNA damage and cell death or cell senescence in stem cells (Table 1). In support of this hypothesis, attenuated DNA damage responses in quiescent stem cells have been observed. Analysis of hematopoietic stem cells from mice deficient in different repair pathways like nucleotide excision repair (XPDTTD mice), telomere maintenance (mTR−/− mice), and non-homologous end joining (Lig4Y288C and Ku80−/− mice) revealed resilience of quiescent stem cells towards damage [33, 34]. Deficiency in any of the repair pathways analyzed did not lead to depletion of stem cell pools with age while their downstream cycling progenitor populations were diminished. Because in these systems, all cells lack efficient repair of DNA damage, the fact that quiescent stem cell pools are not faster depleted with age suggests a reduced DNA damage response in rarely cycling stem cells granting protection from cell death despite the presence of damages. The prospective attenuation of DNA damage responses also results in attenuated DNA repair thus avoidance of stem cell pool depletion comes at an expense of gradual DNA damage accumulation in quiescent cells. In addition, non-dividing cells cannot rely on non-error prone homologous recombination repair but on non-homologous end joining which repairs with low precision. Thus, DNA repair options available for the maintenance of quiescent stem cell pools indirectly contribute to damage accumulation. How do quiescent cells cope with progressive age-related accumulation of genotoxic damage without losing functionality? One key factor in cellular responses to DNA damage is p53 protein, which was previously discussed in connection to symmetric versus asymmetric division of stem cells. In response to DNA damage, p53 protein is stabilized and activated leading to expression of genes that temporarily halt cell cycle to give time for DNA repair (such as p21) [35]. At high levels of genotoxic damage, a distinct phosphorylation pattern instructs p53 to induce cell death via expression of its pro-apoptotic targets (Bax, Noxa, and Puma). Thus, p53 role in cell fate decisions is normally regulated by exact damage levels experienced by the cell. The situation appears to be different in stem cells. In hematopoietic, mammary, and hair follicle bulge stem cells, p53 activity was found to decrease in response to DNA damage [28•, 36] suggesting active downregulation of DNA damage responses by genotoxic stress in adult stem cells. Such downregulation may serve as a protective mechanism which prevents cell death and senescence under high genotoxic pressure experienced by stem cells during aging. Intriguingly, cell death protection occurs also in stem cells that express high p53 levels. Mohrin et al. described increased levels and stabilization of p53 protein in hematopoietic stem cells upon ionizing radiation treatment without prominent detection of cell death in contrast to results obtained for myeloid progenitor cells. The authors postulate that protection from cell death in stem cells may arise from high basal levels of pro-surviving factors along with maintenance of low levels of ROS in the stem cell niche [37•]. Accordingly, a significant increase of >1.5-fold in the basal expression of p53 protein was detected when expression profiles of old versus young mice were compared. However, this increase of p53 was not correlated with differences in the rate of cell death between the two age conditions [15]. It has been found that blocking the downstream targets of p53 (e.g., p21) is protective in stem cells harboring high DNA damage levels due to telomere dysfunction [38]. Interestingly, the strength of DNA damage response and subsequent sensitivity to DNA damage strongly depends on the physical position of stem and progenitor cells within their respective niches [39]. Thus, niche-specific signals are likely to play a significant role in stem cell survival upon DNA damage. In addition to lowering DNA damage responses, the attenuation of genotoxic death in stem cells can be in part attributed to the enhanced expression of pro-survival factors. Multipotent hair follicle bulge stem cells studied in their native niche are profoundly resistant to irradiation-induced apoptosis due to their increased expression of Bcl-2 [36]. Hematopoietic stem cells show enhanced survival upon DNA damage compared to their short-lived myeloid progenitors due to increased expression of the Bcl-2 family of pro-survival genes: Bcl-2, Bcl-xL, Mcl-1, and A1 [37•]. In a separate study, increased expression of Mcl-1 was determined to be essential for regenerative and self-renewal functions of hematopoietic cells [40]. The Bcl-2 family pro-survival proteins function by blocking Bax- and Bak-dependent mitochondrial outer membrane permeabilization, cytochrome-c release, and subsequent activation of the caspase cascade. As a result, cell death is inhibited and mitochondrial integrity is maintained. Importantly, nuclear localization of Bcl-2, Bcl-xL, and Mcl-1 has been suggested to have a role in DNA damage response regulation at site of damage in other cellular contexts [41–43]. Exposure to ionizing radiation in human lung cancer cells enhances nuclear Bcl-2 expression where it directly disrupts the Ku/DNA-PKcs complex, thereby inhibiting error-prone non-homologous end joining repair [41]. Homologous recombination repair pathway was also found to be influenced in DNA damage-treated mouse and human cell lines by Bcl-2, Mcl-1, and Bcl-xL proteins via alterations in post-translational modification of Rad51 [44], interaction with NBS1 [43], and by stabilizing G2/M checkpoint arrest through direct binding with Cdk1(cdc2) [45], respectively. In summary, quiescent stem cells appear to possess regulatory mechanisms which allow them to tolerate sub-lethal amounts of genotoxic damage.
But how is the damage dealt with when stem cells exit quiescence? Recent advances in stem cell analysis revealed some potential hints. The study by Walter et al. demonstrated increased DNA damage-induced death in hematopoietic stem cells exposed to repeated rounds of DNA replication. These data implicate exit from quiescence (and accompanying replication-linked genotoxicity) as the main source of physiological DNA damage in stem cells and suggests that a similar mechanism may underlay stem cell loss during natural aging [46••]. In contrast, two other groups [15, 16••] obtained data indicating rapid repair of DNA damage immediately after cell cycle re-entry in hematopoietic cells. The rapid repair concept goes in line with the option of relying on highly efficient and rapid homologous recombination-based DNA repair pathway which is only active in dividing cells. It needs to be mentioned that all three publications link eventual cell survival to the levels of damage that had accumulated in quiescent cells prior to cell cycle re-entry. Interestingly, no difference in DNA damage response activity was observed when stem cells from old and young mice were compared. Thus, age-related deterioration of stem cells is likely driven by the actual accumulated DNA damage rather than DNA damage response de-regulation.
Global Impact: Systemic Effects of Stem Cell DNA Damage
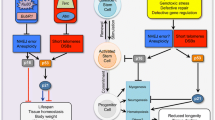
Stem cells play key roles in organ homeostasis and organismal fitness, thus DNA damage in these cells is likely to have a significant systemic impact by causing stem cell dysfunction. Notably, congenital defects in pathways responsible for the maintenance of genome stability often lead to premature aging disorders characterized by a multitude of symptoms including lack of appropriate regeneration—a feature directly dependent on stem cell functionality. Thus, it is feasible that during normal aging, accumulation of DNA damage in stem cells contributes to, or even orchestrates systemic organismal decline. The classical example linking genome instability to prominent stem cell dysfunction is Fanconi anemia (FA) [47]. FA is an autosomal recessive syndrome characterized by bone marrow failure caused by enhanced DNA damage in hematopoietic stem cells driven by lack of DNA inter-strand cross link repair. FA patients succumb to early mortality due to inability to cope with infections and impaired wound healing. Mice suffering from progressive telomere dysfunction due to lack of telomerase develop premature aging symptoms driven by stem cell decline in multiple tissues [48, 49]. Notably telomerase is not expressed in adult somatic cells, thus progeroid features of telomerase deficient mice are solely caused by DNA damage-inflicted stem cell intrinsic failure. Exposure to high doses of ionizing radiation—a so called acute radiation syndrome, manifests features similar to FA and telomerase deficiency—namely failure of the hematopoietic system which is accompanied by strong gastrointestinal dysfunction [50, 51]. Importantly, hematopoiesis and intestinal homeostasis both critically depend on rapid stem cell activity—a process likely hampered by irradiation-driven high levels of DNA damage. Ionizing radiation delivers comparable amounts of DNA damage to all body tissues. While intestine and blood are first to be affected in acute radiation syndrome, other stem cell-dependent systems such as skin and hair also fail at a later time point [52]. Thus, the timeline of DNA-damage driven organ decline is likely linked to normal homeostatic frequency of stem cell cycling in a given tissue. The effect of irradiation likely mimics stem cell dysfunction and organ failure in aging just on a faster scale. In contrast to multi-organ effects of irradiation or telomerase deficiency, congenital DNA repair defects linked to premature aging typically manifest in some tissues and not others. While Cockayne syndrome (block of nucleotide excision repair) strongly manifests in the central nervous system [53], Bloom and Werner syndromes (mutations in RecQ family of genes impairing restart of stalled replication forks) appear to be linked to dysfunction of mesenchymal stem cells [54]. The same is true for Hutchinson Gilford progeria—a very severe premature aging syndrome driven by defects in nuclear lamina [55]. It is feasible that tissue-specific disease manifestations in these cases arise from differential dependence of distinct stem cell types on distinct DNA repair pathways. For example, quiescent hair follicle stem cells rely mostly on non-homologous end joining for repair of double-strand breaks, while actively proliferating intestinal stem cells rely on the homologous recombination pathway. From the studies of premature aging syndromes, it is however clear that DNA damage in stem cells is a strong driver of systemic body decline during diseases and aging (Fig. 1, left panel). Intriguingly, recent data suggests that stem cell DNA damage may not always be harmful. It has been demonstrated that limited DNA damage in stem cell-like germline of C. elegans induces a systemic pro-homeostatic response protecting the whole animal from infections and proteotoxicity [56••]. The protective response involves DNA damage-driven activation of conserved ERK MAP kinase pathway in the germline and induction of the Ubiquitin Proteasome System (UPS) in somatic cells. The UPS activation is triggered by a systemic immune response induced by ERK in the germline and amplified by p38/PMK-1 signaling in the soma. Thus, germline DNA damage orchestrates enhanced systemic fitness of the nematode by boosting protein quality control machinery and improving systemic proteostasis [57, 58]. Interestingly a comparable effect was also observed in a mouse model of sepsis where treatment with anthracyclines—chemotherapy drugs which induce DNA damage, protected animals from lethal septic damage of the lung [59]. Damage protection was dependent on enhanced protein quality control (in this case autophagy) similar to what happens in the nematode upon germline DNA damage. When the germline of C. elegans is damaged, progeny production is temporarily halted letting time for stem cells to repair damage [56••]. Thus, systemic signals which enhance fitness of the whole animal are likely to keep soma alive until reproduction may resume. Given existence of comparable systemic DNA damage response in mammals, it is feasible that limited genotoxic stress in stem cells of at least some tissues instigates systemic protective signals which are meant to reduce organ susceptibility to stress during the time needed for repair processes in stem cells (Fig. 1, right panel). As previously mentioned replacement of dead cells during stress-induced organ damage strongly depends on self-renewal capacity of stem cells. Upon DNA damage, stem cells shift from self-renewal to cell cycle arrest or to p53-driven asymmetric divisions [see chapters 2 and 3] strongly hampering regenerative capacity of the tissue. Thus, systemic protective signals from damaged stem cells may be critical for temporary organ maintenance under stress. Importantly, signaling intermediates found to link stem cell DNA damage to systemic stress tolerance in the nematode are highly conserved in evolution. Induction of innate immune response by DNA damage (UV irradiation) has been demonstrated in the epidermis of Drosophila and mammals [60, 61]. In both cases, MAP kinase signaling was critical for the immune response induction [58]. In the fly DNA damage-inflicted immune activation had strong systemic consequences for the whole body leading to inhibition of insulin signaling and subsequent delay in growth. Such delay could be interpreted as a systemic protective response which reduces homeostatic pressure during time needed for stem cell DNA repair. Systemic consequences of UV exposure-driven immune response in mammals are less understood due to higher complexity of the mammalian system. However, the role of innate immunity in regeneration and homeostasis is well documented in mouse—immune signaling deficient animals show strong impairments in intestinal regeneration upon damage [62–64]. The same is true for repair of skin lesions [65]. Interestingly, both epidermis and intestine not only display links between damage, immunity, and repair but also are highly dependent on stem cells for organ replenishing. Another condition where genome instability is tightly linked to innate immune response is cellular senescence—a terminal outcome of stem cell aging. DNA damage accumulation is the most common driver of cellular senescence. Senescent cells lose the capacity to proliferate and self-renew and switch to secreting a multitude of molecules, mostly pro-inflammatory cytokines [66]. This phenomenon was discovered more than a decade ago and is named senescence associated secretory phenotype (SASP). It is feasible that senescent cells which can no longer contribute to regeneration support organ maintenance by supplying systemic protective signals. The stress protective function of SASP has not so far been confirmed in healthy tissues; however, SASP is a well-known unfortunate helper of tumor growth [67]. Along these lines, it is notable that not only tumor stroma is supported by SASP-generated signals but also cancer stem cells display higher than average incidence of symmetric divisions which strongly contributes to tumor growth [68]. Thus, mechanisms directed at organ protection may once again be highjacked by cancer. It appears feasible that systemic outcomes of stem cell DNA damage are tightly linked to the actual damage load: while high levels of DNA damage caused by acute exposure or lifetime accumulation destroy stem cells and respective organs, limited stem cell DNA damage exerts pro-homeostatic effects (Fig. 1).

Systemic impact of DNA damage in stem cells. The systemic impact of stem cell genotoxic damage is defined by damage strength and load. Strong damages driven by acute genotoxic insults (such as high dose radiation), congenital defects in DNA repair pathways, or accumulation of mild damages with age override protective capacities of stem cells and lead to cell death. Damage-driven stem cell depletion in turn leads to block of regeneration and to functional organ decline. Conversely, mild stem cell DNA damage evokes a systemic immune response leading to systemic induction of protein quality control and elevated fitness. Cells which undergo genotoxic damage-induced senescence are re-programmed to secret pro-inflammatory molecules. These molecules may on the one hand contribute to enhanced fitness via improved protein quality control but on the other hand they promote tumorigenesis
Conclusion
In this review, we discussed differences between adult stem cells of distinct organs in terms of speed of age-related DNA damage accumulation and diverse strategies stem cells use to avoid dying from unbearable damage. The central place of stem cell depletion in organ decline during aging is very clear. In turn, molecular damages play a key role in age-related dysfunction of stem cells. The intriguing concept of damaged stem cells sending pro-homeostatic signals to surrounding tissue(s) still requires extensive investigation. Understanding molecular pathways that underlie enhanced stem cell and tissue functionality in aging will bring us closer to devising pro-homeostatic interventions that delay aging.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95.
Houtkooper RH et al. The metabolic footprint of aging in mice. Sci Rep. 2011;1:134.
Linn S, Kairis M, Holliday R. Decreased fidelity of DNA polymerase activity isolated from aging human fibroblasts. Proc Natl Acad Sci U S A. 1976;73(8):2818–22.
Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190(5):719–29.
Vilchez D, Simic MS, Dillin A. Proteostasis and aging of stem cells. Trends Cell Biol. 2014;24(3):161–70.
Payne BA, Chinnery PF. Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim Biophys Acta. 2015;1847(11):1347–53.
Aguilaniu H et al. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299(5613):1751–3.
Higuchi-Sanabria R. et al., Mitochondrial anchorage and fusion contribute to mitochondrial inheritance and quality control in the budding yeast, Saccharomyces cerevisiae. Mol Biol Cell. 2016;27(5):776–87.
Coelho M et al. Fusion of protein aggregates facilitates asymmetric damage segregation. PLoS Biol. 2014;12(6), e1001886.
Erjavec N et al. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21(19):2410–21.
Rujano MA et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 2006;4(12):e417.
Bufalino MR, DeVeale B, van der Kooy D. The asymmetric segregation of damaged proteins is stem cell-type dependent. J Cell Biol. 2013;201(4):523–30.
Katajisto P et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348(6232):340–3. Describes an age-related asymmetric segregation of mitochondria (13) and altered proteins (14) during division in mammalian stem cells.
Moore DL et al. A mechanism for the segregation of age in mammalian neural stem cells. Science. 2015;349(6254):1334–8. Describes an age-related asymmetric segregation of mitochondria (13) and altered proteins (14) during division in mammalian stem cells.
Beerman I et al. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15(1):37–50.
Moehrle BM et al. Stem cell-specific mechanisms ensure genomic fidelity within HSCs and upon aging of HSCs. Cell Rep. 2015;13(11):2412–24. Shows that stem cell survival and re-entry in cell cycle depends on the levels of DNA damage accumulated during quiescence.
Flach J et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198–202.
Shcheprova Z et al. A mechanism for asymmetric segregation of age during yeast budding. Nature. 2008;454(7205):728–34.
Lark KG, Consigli RA, Minocha HC. Segregation of sister chromatids in mammalian cells. Science. 1966;154(3753):1202–5.
Rocheteau P et al. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148(1–2):112–25.
Waghmare SK, Tumbar T. Adult hair follicle stem cells do not retain the older DNA strands in vivo during normal tissue homeostasis. Chromosom Res. 2013;21(3):203–12.
Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci. 2002;115(Pt 11):2381–8.
Shinin V et al. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol. 2006;8(7):677–87.
Huh YH, Sherley JL. Decreased H3K27 and H3K4 trimethylation on mortal chromosomes in distributed stem cells. Cell Death Dis. 2014;5:e1554.
Talamas JA, Capelson M. Nuclear envelope and genome interactions in cell fate. Front Genet. 2015;6:95.
Cicalese A et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138(6):1083–95.
Merok JR et al. Cosegregation of chromosomes containing immortal DNA strands in cells that cycle with asymmetric stem cell kinetics. Cancer Res. 2002;62(23):6791–5.
Insinga A et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci U S A. 2013;110(10):3931–6. This study provides a link between DNA damage and symmetric self-renewal of stem cells through a p21-dependent downregulation of p53 basal activity.
Yennek S et al. Cell adhesion geometry regulates non-random DNA segregation and asymmetric cell fates in mouse skeletal muscle stem cells. Cell Rep. 2014;7(4):961–70. This study highlights the role of stem cell niche in the asymmetric division of genomic DNA.
Rouleau M et al. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301.
Ying W. Roles of NAD (+), PARP-1, and sirtuins in cell death, ischemic brain injury, and synchrotron radiation X-Ray-induced tissue injury. Scientifica (Cairo). 2013;2013:691251.
Nowsheen S, Yang ES. The intersection between DNA damage response and cell death pathways. Exp Oncol. 2012;34(3):243–54.
Rossi DJ et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9.
Nijnik A et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447(7145):686–90.
Tian XJ et al. A two-step mechanism for cell fate decision by coordination of nuclear and mitochondrial p53 activities. PLoS One. 2012;7(6), e38164.
Sotiropoulou PA et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol. 2010;12(6):572–82.
Mohrin M et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–85. This study postulates that high basal levels of prosurvival factors limit DNA damage-induced cell death in quiescence stem cells.
Choudhury AR et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39(1):99–105.
Tao S et al. Wnt activity and basal niche position sensitize intestinal stem and progenitor cells to DNA damage. EMBO J. 2015;34(5):624–40.
Campbell KJ et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116(17):3197–207.
Wang Q et al. Bcl2 negatively regulates DNA double-strand-break repair through a nonhomologous end-joining pathway. Mol Cell. 2008;29(4):488–98.
Wang J, Beauchemin M, Bertrand R. Phospho-Bcl-x(L)(Ser62) plays a key role at DNA damage-induced G(2) checkpoint. Cell Cycle. 2012;11(11):2159–69.
Jamil S et al. An essential role for MCL-1 in ATR-mediated CHK1 phosphorylation. Mol Biol Cell. 2008;19(8):3212–20.
Saintigny Y et al. A novel role for the Bcl-2 protein family: specific suppression of the RAD51 recombination pathway. EMBO J. 2001;20(10):2596–607.
Schmitt E, Beauchemin M, Bertrand R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene. 2007;26(40):5851–65.
Walter D et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520(7548):549–52. Shows that stem cell survival and re-entry in cell cycle depends on the levels of DNA damage accumulated during quiescence.
Dong H et al. Update of the human and mouse Fanconi anemia genes. Hum Genomics. 2015;9(1):32.
Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464(7288):520–8.
Djojosubroto MW et al. Telomeres and telomerase in aging, regeneration and cancer. Mol Cell. 2003;15(2):164–75.
MacVittie TJ, Farese AM, Jackson 3rd W. The hematopoietic syndrome of the acute radiation syndrome in rhesus macaques: a systematic review of the lethal dose response relationship. Health Phys. 2015;109(5):342–66.
MacVittie TJ et al. The acute gastrointestinal subsyndrome of the acute radiation syndrome: a rhesus macaque model. Health Phys. 2012;103(4):411–26.
Chapel A et al. Mesenchymal stem cells home to injured tissues when co-infused with hematopoietic cells to treat a radiation-induced multi-organ failure syndrome. J Gene Med. 2003;5(12):1028–38.
Weidenheim KM, Dickson DW, Rapin I. Neuropathology of Cockayne syndrome: evidence for impaired development, premature aging, and neurodegeneration. Mech Ageing Dev. 2009;130(9):619–36.
Cheung HH et al. Telomerase protects Werner syndrome lineage-specific stem cells from premature aging. Stem Cell Rep. 2014;2(4):534–46.
Zhang J et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8(1):31–45.
Ermolaeva MA et al. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature. 2013;501(7467):416–20. This study provides evidence that limited genotoxic stress in stem cells triggers a systemic immune response which allows animal protection against infections and proteotoxicity.
Ermolaeva M, Schumacher B. The innate immune system as mediator of systemic DNA damage responses. Commun Integr Biol. 2013;6(6), e26926.
Ermolaeva MA, Schumacher B. Systemic DNA damage responses: organismal adaptations to genome instability. Trends Genet. 2014;30(3):95–102.
Figueiredo N et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity. 2013;39(5):874–84.
Karpac J, Younger A, Jasper H. Dynamic coordination of innate immune signaling and insulin signaling regulates systemic responses to localized DNA damage. Dev Cell. 2011;20(6):841–54.
Clydesdale GJ, Dandie GW, Muller HK. Ultraviolet light induced injury: immunological and inflammatory effects. Immunol Cell Biol. 2001;79(6):547–68.
Rakoff-Nahoum S et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118(2):229–41.
Fukata M et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288(5):G1055–65.
Rose 2nd WA, Sakamoto K, Leifer CA. TLR9 is important for protection against intestinal damage and for intestinal repair. Sci Rep. 2012;2:574.
Lin ZQ et al. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol. 2003;73(6):713–21.
Rodier F et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–9.
Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67(7):3117–26.
Gomez-Lopez S, Lerner RG, Petritsch C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell Mol Life Sci. 2014;71(4):575–97.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Lilia Espada and Maria A. Ermolaeva declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Age-related Stem Cell Modifiers
Rights and permissions
About this article

Cite this article
Espada, L., Ermolaeva, M.A. DNA Damage as a Critical Factor of Stem Cell Aging and Organ Homeostasis. Curr Stem Cell Rep 2, 290–298 (2016). https://doi.org/10.1007/s40778-016-0052-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-016-0052-6