
Abstract
Computational modelling is a cornerstone of Comprehensive In Vitro Proarrhythmia Assay and is re-increasingly being used in drug development. Electrophysiological effects of drug-drug interactions can be predicted in silico, e.g. with the use of in vitro cardiac ion channel data, PK profiles and human ventricular cardiomyocyte models. There are, however, several approaches with different assumptions used to assess the combined effect of multiple drugs, and there is no agreed standard interaction model. The aim of this study was to assess whether the choice of the drug-drug interaction (DDI) model (Bliss independence, Loewe additivity, or simple sum) influences the results of QT interval simulation trial. The Simcyp Simulator version 12.1 (Simcyp Ltd. [part of Certara], Sheffield, UK) and Cardiac Safety Simulator 2.0 (Simcyp Ltd. [part of Certara], Sheffield, UK) were used to simulate results of 8 virtual trials mimicking clinical studies and generate individual QTc data. The combined effect of inhibitory actions of drugs which were given simultaneously was calculated with use of three different interaction models. The PD effect of DDI was assessed and the differences between mean observed and mean predicted ΔQTcB values for terfenadine interactions were not statistically significant in all but one cases. Differences between the three DDI models are not statistically significant, implying that the choice of the DDI model, in the case of lack of synergy or antagonism, is irrelevant to the average predicted effect at the clinical level. However, in some cases, it can influence the verdict on combinatorial therapy safety for individual patients.
Similar content being viewed by others

Introduction
Polypharmacy is a fact of life in the clinic. Medicine in general and pharmacotherapy, in particular, take advantages of combination therapies in many diseases, e.g. hypertension, diabetes, asthma or cancer. When intended, drug interactions resulting from drug combinations maximize the efficacy of a therapy and minimize adverse effects. On the other hand, however, in the majority of cases, simultaneous use of several active compounds complicates the pharmacotherapy and may lead to increased risk of health- or life-threatening drug-drug interactions (DDI). The mechanism of interaction can be specific for the pharmacokinetics (PK) of the combined drugs as well as their pharmacodynamic (PD) activity or combine both areas—PK/PD. PK interaction can occur at any level of the absorption, distribution, metabolism, and excretion (ADME). The detailed processes vary for a different element of drugs pharmacokinetics [26]. There are plethora of potential interaction mechanisms at the level of drug action. Pharmacodynamic interactions occur when a precipitant drug alters the clinical effects of the object drug at its site of action [4]. This alteration is caused by modification of the environment which finally results in increasing or decreasing the expected clinical effects. One of the potential harmful effects of DDI is the QT interval prolongation and torsade de pointes (TdP) arrhythmia.
DDIs are studied in vitro or in vivo during clinical trials. However, many of potential adverse effects being a consequence of DDI are not revealed until the drug is marketed because of, among other issues, infeasibility of experimental testing of all possible drug combinations. Computational approaches offer an advantage in DDIs evaluation as it is possible to explore wide space of drug combinations using various types of drug data (e.g. chemical structure, physico-chemical properties, information about targets) [3, 12, 24, 27, 33, 34, 36, 37].
In silico modelling is a cornerstone of CiPA (Comprehensive in vitro Proarrhythmia Assay) initiative and currently witnessed proarhythmia assessment paradigm shift. To incorporate DDIs into proarrhythmia modelling, it is necessary to assess the combined effect of multiple drugs on channel or electrocardiogram (ECG) level. There are several approaches used to assess the combined effect of multiple drugs [8]. Proposed DDI models differ in their assumptions, and there is a lack of consensus on the standard interaction model [31]. The Loewe additivity [23] and Bliss independence [2] models are widely used and applied to various problems [11]. The main assumption of the Loewe model is that all the interacting compounds in a combination act on the same target or binding site through the same mechanism while Bliss model assumes exactly the opposite, i.e. drugs in a combination act independently, yet they can perturb their individual responses. Both the models assume the lack of synergistic and antagonistic interaction between drugs. In such situation, there is also the possibility to consider the effect of interaction as a simple sum of the effects of single compounds up to some physiologically feasible maximum level.
The aim of this study was to assess whether the choice of the DDI model influences the results of QT interval simulation trial and which of the applied interaction models generates the results closest to the observed values. Terfenadine was chosen as an exemplary case as there are published results of several clinical studies where different QT prolonging drugs were administered concomitantly with terfenadine.
Methods
The Simcyp Simulator version 14.1 (Simcyp Ltd. [part of Certara], Sheffield, UK) and Cardiac Safety Simulator 2.0 [10] were used to simulate results of eight virtual trials mimicking clinical studies. It is worth mentioning that these eight trials cover all information on that topic in the publicly available scientific sources [35]. Simcyp platform enabled to estimate drug exposure resulting from DDIs between terfenadine and co-administrated metabolic inhibitors (clarithromycin, erythromycin, fluconazole, fluoxetine, itraconazole, ketoconazole, and paroxetine). The simulations of the pharmacokinetics of terfenadine alone and in the presence of metabolic inhibitor were performed using full physiologically based pharmacokinetic (PBPK) model to allow to simulate the heart tissue concentrations. Pharmacodynamic effects of DDIs in the population of patients were simulated with the use of Cardiac Safety Simulator.
Both PK and PD components of DDI simulation were designed to mimic all settings of the above mentioned clinical trials in terms of demographic, physiological and genetic characteristics of the observed populations. Each study was simulated 10 times to account for the inter-study variability as described previously [35].
Three different interaction models were applied to calculate the combined effect of inhibitory actions of drugs which were given simultaneously, i.e. (1) simple arithmetic sum with a maximal inhibition limit of 1 (all channels blocked in 100 %), (2) Bliss independence, and (3) Loewe additivity to assess which of the applied interaction models generates the results closest to the observed values.
-
Equation 1.
Simple sum model.
-
Equation 2.
Bliss independence model [2].
-
Equation 3.
Loewe additivity model [23].
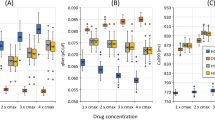
Figure 1 presents expected differences in DDI effect predicted by three different models assuming the same concentrations and equal potencies of interacting drugs (A, B).

Resultant channel inhibition for 3 DDI models. Equal concentrations and potencies assumed; [DRUG A] = [DRUG B]; IC50 = 1 μM, n = 1. S—simple sum model; B—Bliss model; L—Loewe model
For all tested interaction models, the maximum current inhibition was set to 1 (100 %).
The measure of distance (D) was calculated as follows:
where:
- Δ:
-
QT interval change
- T:
-
terfenadine
- I:
-
inhibitor
- pred:
-
predicted value
- obs:
-
observed value
It was possible to compare the average values only as the observations for individual patients were not reported in the literature. Predictions for individual patients were analysed separately to determine the relevance of the model choice for the drug cardiac safety assessment. Safety threshold for which assumed DDI model gives results which are of relevance to conclude on potential health-threatening was defined as 5 ms of the heart rate corrected QT prolongation. A mean QTc interval prolongation of 5 ms with the upper limit of 95 % confidence interval above 10 ms is defined as threshold pharmacologic effect of a drug on cardiac repolarization and the threshold level of regulatory concern (ICH [20]). For the sake of this analysis, the case was defined as a situation where using different interaction model would change the decision about meeting the above-defined threshold. The 5 ms threshold can be recognized as very strict, especially considering the expected population variability; however, this was used to mimic the ICH E14 suggested value.
Results
The simulated and the reported clinical values of area under the curves (AUCs) and maximum plasma concentrations (Cmax) are given in Table 1.
Table 2 presents results of simulation QT studies and compares them with the reported clinical values. Distance is the measure indicating goodness of fit of the predicted ΔQTc values as compared against the observed one.
Predicted with the use of three different interaction models and observed average changes of QTcB interval lengths (baseline subtracted) resulting from concomitant administration of terfenadine and an inhibitor are presented on Fig. 2.

Baseline subtracted changes of QTcB interval—predicted vs. observed
Percentage of cases meeting the criterion of reaching the safety threshold (5 ms increase for QTcB) for all analysed models are presented in Table 3.
Discussion and Conclusions
The early assessment of cardiac safety liabilities is essential to advance novel drug candidates confidently. Apart from being early, the screening system should also be efficient and cost-effective. Detection of the drug-induced electrophysiological toxic effects is, therefore, crucial. The new paradigm of cardiac safety testing using in vitro human ion channel assays, human-based in silico reconstructions and human stem cell-derived cardiomyocytes has been recently discussed [9]. One of the characteristics of all safety testing systems is their focus on the single chemical entity. Although, as it was described before in the clinical settings, the situation where a single drug is given is relatively rare and polypharmacy is very common. Therefore, system for the safety assessment of the drug combinations would be needed. From the above-listed systems and methods human-based in silico reconstructions of cardiac electrophysiology look best suited for such need. If properly validated, computer simulations with the use of biophysically detailed models can offer precise information about the clinical consequences of drug combinations without running clinical trials, especially when combined with the already commonly used PBPK models used for the exposure assessment.
In the current study, the pseudoECG signals were simulated for terfenadine alone and in the presence of seven different compounds which are inhibitors of terfenadine metabolism as well as known QT prolonging drugs. The pharmacokinetic component of the DDI was modelled and simulated with the use of Simcyp Simulator. The predicted AUC and Cmax values for terfenadine alone and with the concomitantly given inhibitors were close to the clinically observed values as presented in the recent publication [35]. Predicted terfenadine and inhibitor exposure profiles were utilized to calculate ion channels inhibition and current changes (IKr, IKs, INa, ICa) resulting from drug combination. The latter was done assuming three models of DDI effect. Current specific, maximal ion channel conductances expressed in the ten Tusscher human ventricular cardiomyocyte model were modified by the calculated total inhibition of ion currents. The inhibition was drug concentration dependent, under the assumptions that the in vitro measured activity equals to in vivo situation and that the maximum inhibition cannot exceed 100 %. The ΔQTcB was calculated on the basis of simulated pseudoECG signals for baseline (no drug), terfenadine alone, and terfenadine plus inhibitor scenarios.
The prediction of drug-drug interaction at the PD level was assessed with the use of statistical methods. The differences between mean observed and mean predicted ΔQTcB values for terfenadine interactions were not statistically significant (Welch t test) for most of the studies (except for ketoconazole study). Some discrepancies between predictions and clinical observations may arise from the in vitro data variability, small populations of individuals included to the clinical trials, or ΔQTcB calculation method (different definition of baseline).
The hERG channel is the main site of PD interaction between terfenadine and perpetrating drugs used in this study. The Loewe and Bliss DDI model assume the same or different binding sites within channel pore for simultaneously acting channel blockers. Since erythromycin [5, 7] and possibly clarithromycin [6] bind to the external domain of hERG channel protein, while terfenadine [21], fluoxetine [14, 28], fluconazole [13], ketoconazole [29, 30, 32], paroxetine [22] and presumably itraconazole bind to internal domain of hERG channel pore, the Bliss and Loewe model, respectively, should perform better for those groups of drugs. This is not the case according to the results obtained it this study. As indicated in Table 2, in almost all cases, simple sum method yield the closest results to those observed in clinical trials. However, differences between the three DDI models are not statistically significant, implying that the choice of the DDI model, in case of lack of synergy or antagonism, is irrelevant to the average predicted effect at the clinical level.
In the thorough QT (TQT) studies, threshold of regulatory concern for QT prolongation is set as 5 ms (ICH [20]). The percentage of individual patient records, where the difference between the models predictions was at least 5 ms ranged from 0 to 78 % depending on the perpetrating drug. However, only cases where the choice of the model could be of relevance for the conclusions drawn, i.e. where the models predictions differ and put the QTc change on the opposite sites of the safety threshold, can be interpreted as important difference. As shown in Table 3, the percentage of such cases is low. It can be concluded that the model choice can influence the verdict on combinatorial therapy safety for individual patients, but it has a negligible effect at the level of population.
References
Bergstrom RF, Goldberg MJ, Cerimele BJ, Hatcher BL. Assessment of the potential for a pharmacokinetic interaction between fluoxetine and terfenadine. Clin Pharmacol Ther. 1997;62(6):643–51.
Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26(3):585–615.
Bulusu KC, Guha R, Mason DJ, Lewis RPI, Muratov E, Motamedi YK, Cokol M, Bender A. Modelling of compound combination effects and applications to efficacy and toxicity: state-of-the-art, challenges and perspectives. Drug Discov Today. 2016;21(2):225–38. doi:10.1016/j.drudis.2015.09.003.
Corrie K, Hardman JG. Mechanisms of drug interactions: pharmacodynamics and pharmacokinetics. Anaesthesia Intensive Care Med, Neonatal / Pharmacol. 2011;12(4):156–9. doi:10.1016/j.mpaic.2010.12.008.
Crumb WJ. Allosteric effects of erythromycin pretreatment on thioridazine block of hERG potassium channels. Br J Pharmacol. 2014;171(7):1668–75. doi:10.1111/bph.12575.
Du, Chunyun, Aziza El Harchi, Henggui Zhang, and Jules C Hancox. ‘Modification by KCNE1 variants of the hERG potassium channel response to premature stimulation and to pharmacological inhibition’. Physiol Reports. 2013;1(6). doi:10.1002/phy2.175.
Duncan RS, Ridley JM, Dempsey CE, Leishman DJ, Leaney JL, Hancox JC, Witchel HJ. Erythromycin block of the HERG K+ channel: accessibility to F656 and Y652. Biochem Biophys Res Commun. 2006;341(2):500–6. doi:10.1016/j.bbrc.2006.01.008.
Foucquier, Julie, and Mickael Guedj. ‘Analysis of Drug Combinations: Current Methodological Landscape’. Pharmacol Res Perspect. 2015;3(3). doi:10.1002/prp2.149.
Gintant G, Sager PT, Stockbridge N. ‘evolution of strategies to improve preclinical cardiac safety testing’. Nature Reviews. Drug Discovery. 2016;15(7):457–71. doi:10.1038/nrd.2015.34.
Glinka A, Polak S. QTc modification after risperidone administration—insight into the mechanism of action with use of the modeling and simulation at the population level approach. Toxicol Mech Methods. 2015;25(4):279–86. doi:10.3109/15376516.2015.1025346.
Goldoni M, Johansson C. A mathematical approach to study combined effects of toxicants in vitro: evaluation of the bliss independence criterion and the Loewe additivity model. Toxicol in Vitro. 2007;21(5):759–69. doi:10.1016/j.tiv.2007.03.003.
Gottlieb A, Stein GY, Oron Y, Ruppin E, Sharan R. INDI: a computational framework for inferring drug interactions and their associated recommendations. Mol Syst Biol. 2012;8:592. doi:10.1038/msb.2012.26.
Han S, Zhang Y, Chen Q, Duan Y, Zheng T, Hu X, Zhang Z, Zhang L. Fluconazole inhibits hERG K(+) channel by direct block and disruption of protein trafficking. Eur J Pharmacol. 2011;650(1):138–44. doi:10.1016/j.ejphar.2010.10.010.
Hancox JC, Mitcheson JS. Combined hERG channel inhibition and disruption of trafficking in drug-induced long QT syndrome by fluoxetine: a case-study in cardiac safety pharmacology. Br J Pharmacol. 2006;149(5):457–9. doi:10.1038/sj.bjp.0706890.
Honig PK, Woosley RL, Zamani K, Conner DP, Cantilena Jr LR. Changes in the pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine with concomitant administration of erythromycin. Clin Pharmacol Ther. 1992;52(3):231–8.
Honig PK, Worham DC, Zamani K, Mullin JC, Conner DP, Cantilena LR. The effect of fluconazole on the steady-state pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine in humans. Clin Pharmacol Ther. 1993a;53(6):630–6.
Honig PK, Wortham DC, Hull R, Zamani K, Smith JE, Cantilena LR. Itraconazole affects single-dose terfenadine pharmacokinetics and cardiac repolarization pharmacodynamics. J Clin Pharmacol. 1993b;33(12):1201–6.
Honig PK, Wortham DC, Zamani K, Conner DP, Mullin JC, Cantilena LR. Terfenadine-ketoconazole interaction. Pharmacokinetic and electrocardiographic consequences. JAMA. 1993c;269(12):1513–8.
Honig PK, Wortham DC, Zamani K, Cantilena LR. Comparison of the effect of the macrolide antibiotics erythromycin, clarithromycin and azithromycin on terfenadine steady-state pharmacokinetics and electrocardiographic parameters. Drug Investigation. 1994;7(3):148–56.
ICH 2005. ‘Guidance for Industry. E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs’, no. Journal Article. 2005.
Kamiya K, Niwa R, Morishima M, Honjo H, Sanguinetti MC. Molecular determinants of hERG channel block by terfenadine and cisapride. J Pharmacol Sci. 2008;108(3):301–7.
Lee SH, Sung MJ, Lee HM, Chu D, Hahn SJ, Jo S-H, Choe H, Choi BH. Blockade of HERG human K+ channels by the antidepressant drug paroxetine. Biol Pharm Bull. 2014;37(9):1495–504.
Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953;3(6):285–90.
Luo, Heng, Ping Zhang, Hui Huang, Jialiang Huang, Emily Kao, Leming Shi, Lin He, and Lun Yang. ‘DDI-CPI, a server that predicts drug-drug interactions through implementing the chemical-protein interactome’. Nucleic Acids Res. 2014;42(Web Server issue): W46–52. doi:10.1093/nar/gku433.
Martin DE, Zussman BD, Everitt DE, Benincosa LJ, Etheredge RC, Jorkasky DK. Paroxetine does not affect the cardiac safety and pharmacokinetics of terfenadine in healthy adult men. J Clin Psychopharmacol. 1997;17(6):451–9.
Palleria C, Di Paolo A, Giofrè C, Caglioti C, Leuzzi G, Siniscalchi A, De Sarro G, Gallelli L. Pharmacokinetic drug-drug interaction and their implication in clinical management. J Res Med Sci: Off J Isfahan Univ Med Sci. 2013;18(7):601–10.
Park K, Kim D, Ha S, Lee D. Predicting pharmacodynamic drug-drug interactions through signaling propagation interference on protein-protein interaction networks. PLoS One. 2015;10(10):e0140816. doi:10.1371/journal.pone.0140816.
Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, Holzem KM, et al. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149(5):481–9. doi:10.1038/sj.bjp.0706892.
Ridley JM, Milnes JT, Duncan RS, McPate MJ, James AF, Witchel HJ, Hancox JC. Inhibition of the HERG K+ channel by the antifungal drug ketoconazole depends on channel gating and involves the S6 residue F656. FEBS Lett. 2006;580(8):1999–2005.
Takemasa H, Nagatomo T, Abe H, Kawakami K, Igarashi T, Tsurugi T, Kabashima N, et al. Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome. Br J Pharmacol. 2008;153(3):439–47. doi:10.1038/sj.bjp.0707537.
Tang, Jing, Krister Wennerberg, and Tero Aittokallio. ‘What is synergy? The Saariselkä agreement revisited’. Front Pharmacol. 2015;6(September). doi:10.3389/fphar.2015.00181.
Thurner P, Stary-Weinzinger A, Gafar H, Gawali VS, Kudlacek O, Zezula J, Hilber K, Boehm S, Sandtner W, Koenig X. Mechanism of hERG channel block by the psychoactive indole alkaloid ibogaine. J Pharmacol Exp Ther. 2014;348(2):346–58. doi:10.1124/jpet.113.209643.
Vilar S, Harpaz R, Uriarte E, Santana L, Rabadan R, Friedman C. Drug-drug interaction through molecular structure similarity analysis. J Am Med Inform Assoc: JAMIA. 2012;19(6):1066–74. doi:10.1136/amiajnl-2012-000935.
Vilar S, Uriarte E, Santana L, Lorberbaum T, Hripcsak G, Friedman C, Tatonetti NP. Similarity-based modeling in large-scale prediction of drug-drug interactions. Nat Protoc. 2014;9(9):2147–63. doi:10.1038/nprot.2014.151.
Wiśniowska B, Polak S. Virtual clinical trial toward polytherapy safety assessment: combination of physiologically based pharmacokinetic/pharmacodynamic-based modeling and simulation approach with drug-drug interactions involving terfenadine as an example. J Pharm Sci. 2016;105(11):3415–24. doi:10.1016/j.xphs.2016.08.002.
Zhang L, Zhang Y(D), Zhao P, Huang S-M. Predicting drug–drug interactions: an FDA perspective. AAPS J. 2009;11(2):300–6. doi:10.1208/s12248-009-9106-3.
Zhang, Ping, Fei Wang, Jianying Hu, and Robert Sorrentino. ‘Label propagation prediction of drug-drug interactions based on clinical side effects’. Sci Reports 2015;5(July): 12339. doi:10.1038/srep12339.
Acknowledgments
This work was supported by the Polish National Science Center [DEC 2013/11/D/NZ7/01583].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
SP is an employee of Simcyp (part of Certara).
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pharmacometrics
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wiśniowska, B., Polak, S. The Role of Interaction Model in Simulation of Drug Interactions and QT Prolongation. Curr Pharmacol Rep 2, 339–344 (2016). https://doi.org/10.1007/s40495-016-0075-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-016-0075-9