
Abstract
In recent years, many pre-clinical studies have tested gene therapy approaches as possible treatments for epilepsy, following the idea that they may provide an alternative to conventional pharmacological and surgical options. Multiple gene therapy approaches have been developed, including those based on anti-sense oligonucleotides, RNA interference, and viral vectors. In this opinion article, we focus on translational issues related to viral vector-mediated gene therapy for epilepsy. Research has advanced dramatically in addressing issues like viral vector optimization, target identification, strategies of gene expression, editing or regulation, and safety. Some of these pre-clinically validated potential gene therapies are now being tested in clinical trials, in patients with genetic or focal forms of drug-resistant epilepsy. Here, we discuss the ongoing translational research and the advancements that are needed and expected in the near future. We then describe the clinical trials in the pipeline and the further challenges that will need to be addressed at the clinical and economic levels. Our optimistic view is that all these issues and challenges can be overcome, and that gene therapy approaches for epilepsy will soon become a clinical reality.
Similar content being viewed by others


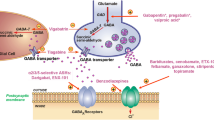
Multiple gene therapy approaches are currently being tested for drug-resistant epilepsies, in particular those based on anti-sense oligonucleotides and on viral vectors. |
Two phase I/II viral vector-based gene therapy clinical studies that target focal and genetic forms of epilepsy are now starting. |
Viral vector-based gene therapy approaches have reached high levels of optimization but refinement is still required. |
1 Epilepsies
The term “epilepsy” refers to a collection of diseases characterized by the enduring predisposition to generate spontaneous unpredictable seizures [1]. Although seizures are the main sign of the disorder, their origin, semiology, frequency, and severity can be extremely heterogeneous, reflecting many possible etiologies [2]. Seizures can be generalized, when the electrical activity occurs in bilaterally distributed networks, or focal, when activity is limited to one hemisphere. Epilepsies associated with generalized seizures are often caused by a genetic defect, whereas epilepsies with focal seizures generally result from a lesion in a specific brain region. More than 30% of the patients are drug resistant [3, 4] and may experience a wide range of comorbidities, mostly psychiatric and cognitive, as well as social complications. In addition, people with epilepsy also have an increased risk of premature death mainly due to sudden unexpected death in epilepsy [5].
There are many unmet clinical needs in this disorder, in particular [6], the development of treatments for drug-resistant epilepsies, the improvement of tolerability of treatments, identification of disease-modifying treatments that prevent or attenuate the development of epilepsy in at-risk individuals (e.g., children experiencing febrile seizures, individuals with a brain trauma, and individuals affected by brain tumors, stroke, or Alzheimer’s disease), and the development of treatments capable of preventing or ameliorating the common comorbidities that contribute to disability. The introduction of new drugs into clinical practice over the past two decades has not substantially changed the situation [7]. Epilepsy surgery, consisting of the removal of the abnormal part of the brain, is effective for carefully selected patients with drug-resistant focal epilepsies [8] but, unfortunately, this option can be offered only to a small number of patients because many epilepsies do not have a focal origin and because of the risk of affecting important brain functions, for example, resecting the eloquent cortex. In addition, epilepsy surgery centers require highly specialized personnel and are very expensive, strongly limiting access to treatment for many of those who may benefit, especially in low-income countries. Evidence of efficacy and safety of other approaches, such as vagal nerve stimulation, deep brain and cortical stimulation, or a ketogenic diet, is still insufficient [9, 10]. In sum, the identification of alternative, more effective treatment options for difficult-to-treat epilepsies is highly urgent.
2 Might Gene Therapy Become an Option for Epilepsy Treatment?
There is growing pre-clinical evidence to indicate that gene therapy is a suitable approach to treat a large population of patients with epilepsy that have no therapeutic alternatives. The concept of gene therapy has evolved over the years, and currently includes not only the introduction of healthy variants of genes in cells, but also the modulation of existing gene activity and gene editing. These goals may be pursued using different cargo delivery methods (nanoparticles, viruses, ribonucleoprotein complexes), different approaches (patient-derived cells treated ex vivo, direct in vivo injection), and different gene modulation strategies (CRISPR-Cas9, anti-sense oligonucleotides, microRNAs) [11]. Altogether, gene therapy is now emerging as a promising therapeutic strategy for many diseases, including diseases that may not have a genetic cause, such as Parkinson’s disease and Alzheimer’s disease [12].
Gene therapy approaches for epilepsy can be divided into two broad categories: those targeting the gene defect, for genetic forms of the disease; and those targeting mechanism(s) of seizure generation [13]. In both cases, different gene therapy approaches have been developed, based on anti-sense oligonucleotides, RNA interference, and viral vectors. In this opinion paper, while referring the reader to other articles discussing other approaches [13, 14], we focus on in vivo gene therapies mediated by viral vectors. For such an approach to be successful, many issues should be considered and addressed, for example, optimization of the natural ability of viruses to transfer genetic material into target cells; production of clinical-grade vectors; targeting of specific cell populations in a highly heterogeneous environment; long-lasting and modulated expression of therapeutic genes; and safety. In the following paragraphs, we discuss recent developments and future prospects in these many respects, before describing the first clinical trials that are now ready to start.
3 Viral Vectors
To be effective, gene therapy for epilepsy needs to achieve stable, highly regulated, and safe gene expression in the brain. Viral vectors based on different viruses [adenovirus, adeno-associated virus (AAV), lentiviruses (LV), herpes simplex virus] have been shown to represent valid options to achieve these requirements [12, 15, 16]. Among these, AAV- and LV-based vectors are emerging as the most promising.
Recombinant AAV vectors seem particularly suited to central nervous system (CNS) gene therapy because of their low pathogenicity, high delivery efficiency, and specific tissue or cell tropism [17]. Many different recombinant AAV vector-based gene therapy products are already approved for use in patients with lipoprotein lipase deficiency, Leber’s congenital amaurosis, spinal muscular atrophy, and Duchenne muscular dystrophy [18, 19]. Recombinant AAV vectors ensure long-lasting transgene expression and a broad spread in the parenchyma after direct injection in the brain [20], which makes them good candidates to target focal epileptogenic areas. In addition, the discovery of new AAV capsids, such as AAVrh.8, AAVrh.10, and AAV9, which are able to cross the blood–brain barrier (BBB), hold the promise of application to genetic forms of epilepsy that may require targeting the entire brain [21, 22]. The downside of AAV vectors is the very limited cargo capacity and, therefore, efforts are directed to addressing this issue [23].
Notwithstanding their limitations, for example, the inability to cross the BBB and the limited spread from the site of injection in the brain tissue [24], LV vectors remain a promising strategy to treat neurological diseases because of their larger cargo capacity and their efficiency in providing permanent expression of transgenes in non-dividing cells such as neurons [25]. They are currently studied for gene therapy of neurometabolic diseases that can display epileptic symptoms [26, 27]. In addition, their ability to provide rapid, stable, and spatially restricted transgene expression makes them a very interesting tool for focal epilepsies [28]. One issue that must be addressed relates to the fact that LVs insert their DNA in the host cell genome, with the consequent risk of mutagenesis. However, this risk is highly attenuated with last-generation LV vectors [29, 30].
Independent of the type of viral backbone, one common hurdle is the production of clinical-grade vectors. High yield, concentration, and purity are necessary attributes of clinical-grade vectors, owing to the large amounts of viral particles that are needed for transducing relatively broad brain areas or to overcome the BBB. Improvements in the scalability of recombinant AAV vector production can be achieved through different strategies [31,32,33,34,35]. There is still room for improvement, as some of these issues (e.g., DNA contamination or non-infective particles) remain a fundamental challenge. Describing all the issues related to large-scale production of vectors goes beyond the scope of this paper. Nonetheless, it will be very important to overcome them, as the very high manufacturing cost of gene therapy products has a huge impact on their affordability [20].
4 Targets and Strategies
As mentioned above, gene therapy strategies for epilepsy may aim at targeting the genetic defect or the mechanism(s) of seizure generation. The former strategy may be the most obvious for genetic forms of epilepsy. In these cases, however, the genetic defect affects widespread populations of cells in the CNS, and the gene therapy vector should therefore be able to cross the BBB or diffuse broadly after injection in the cerebrospinal fluid, a goal that can be achieved using specific AAV serotypes [36, 37]. In the epilepsy field, promising results have been obtained for Dravet syndrome (DS) [38], a severe infantile epilepsy syndrome that affects approximately 2.5 in 100,000 children [39]. Dravet syndrome is a developmental epileptic encephalopathy that is most often caused by monogenic loss-of-function variants of the SCN1A gene, which encodes the alpha subunit of the voltage-gated type I sodium channel (NaV1.1). This leads to impaired activity of neurons, primarily inhibitory GABAergic neurons [40]. Recent data suggest that DS may be treated efficiently with a AAV gene therapy approach, ETX101. ETX101 uses a non-replicating recombinant AAV9 in which a GABAergic regulatory element ensures selective expression in GABAergic neurons of an engineered transcription factor that increases SCN1A gene transcription [38]. A single injection of ETX101 in a mouse model of DS induced increased SCN1A gene activity and NaV1.1 protein expression in brain GABA neurons, as well as a significant reduction in spontaneous seizures and improved long-term survival [38].
The alternative option is to target the mechanisms underlying the generation of seizures [41, 42], which has mainly been tested via a direct injection of vectors in the epileptogenic region, in models of focal epilepsies. In this respect, several strategies have been proposed and characterized based on the transfer in the epileptogenic area of genes that modify cell function and control hyperexcitability, such as ion channels [43], neurotransmitters [44, 45], neurotrophic factors [46], or receptors [47]. Other strategies that have been tested include the transfer of genes encoding proteins that render the cell sensitive to specific drugs (chemogenetics [48]) or to light stimulation (optogenetics [49]). A combinatory gene therapy may be useful to disrupt more than one mechanism of seizure generation. One example is the use of viral vectors to locally supplement a combination of the neurotransmitter neuropeptide Y (NPY) with its receptor Y2, which proved superior compared with NPY alone [47, 50]. Finally, recent studies explored the possibility of using gene editing technologies, which proved very promising both in models of genetic and focal lesional epilepsy [51, 52].
5 Control of Transgene Expression
Finely tuning transgene expression in terms of cell specificity, temporal (on-demand) expression, and levels of expression would increase safety and efficacy. First, transgene expression in specific cell populations is often essential for epilepsy gene therapy. For example, anti-seizure effects may be obtained by increasing the strength of inhibitory signals or reducing that of excitatory signals, but these effects depend on the cell population expressing the transgene, that is, selective inhibition of excitatory, but not inhibitory, neurons can produce anti-seizure effects [12]. Targeting the therapeutic intervention on specific brain cell populations may be obtained by various means, for example, driving transgene expression by cell-specific promoters [16]; incorporating tandem repeats of artificial microRNA target sites into the 3′ UTR of the transgene expression cassette, which leads to degradation of transgene mRNA in cells expressing the corresponding microRNA [53]; and using specific AAV serotypes [54]. Although none of these systems is perfect, they may be combined in an attempt to achieve a high degree of cell specificity.
In addition, all protocols that have been experimentally tested thus far have applied gene therapy at a single dose, and this was irreversible, with a fixed gene expression level and the consequent risk of under-dosing or over-dosing. This way, expression of the transgene could not be stopped or modified according to the specific patient’s needs. Regulated or “on-demand” inducible systems would therefore be desirable. Controlled transgene expression can be obtained in multiple ways [16], for example, by using inducible promoters, obtained by incorporating in the vector (or in a separate vector) a cassette driving the constitutive expression of a transcription factor (transactivator) able to activate or block transgene expression depending on the availability of a molecule that is administered systemically (e.g., doxycycline, rapamycin); or by using promoters activated only when neurons are hyper-excited [55].
Finally, self-regulation or transgene expression may be obtained by engineering vectors to respond to internal environmental cues, such as inflammation or hypoxia [56, 57]. These systems are not yet fully developed. Externally regulated systems (inducible promoters, optogenetics, DREADDs) seem more attractive, but entail issues related to the safety of the transgene expression inducer (drug or device) and to the potential leakage of the system in the absence of the inducer. Moreover, the expression of non-self-proteins implies potential immunogenicity (see below).
Last but not least, all systems described in this section require expanding the size of the expression cassette and/or expressing multiple genes. As discussed above, this implies several technical problems, such as limited payload capacity of the viral genome for AAVs and positional effects due to varying gene positions in multi-cistronic constructs [58, 59], particularly for gene products that are delivered to different subcellular compartments or display post-translational modifications [60, 61].
6 Toxicity and Immunogenicity
Viral vector-mediated gene therapy is expected to cause dose-dependent adverse effects [62,63,64,65,66]. However, little evidence regarding this issue is as yet available for CNS gene therapy, because almost all pre-clinical studies are conducted with single doses. This complicates the translation to human studies that are generally conducted with single doses (even if dose-escalating designs were employed in first-in-human gene therapy studies in patients with Parkinson’s disease [67,68,69,70]). Experimental designs including dose finding would be useful in future pre-clinical and, if ethically acceptable, first-in-human clinical studies of gene therapy for epilepsy.
Intracranial administration of vectors into the CNS, while being less prone to evoke immune reactions (see below), may entail other risks, in particular in people with epilepsy because the vectors themselves may cause neuroinflammation [71,72,73] and thereby facilitate seizures. Pre-clinical studies in neurodegenerative disorders reported that infiltration by innate immune cells interferes with AAV vectors and compromises the gene therapy effect after intracerebroventricular, intra-cisterna magna, intrathecal, and intraparenchymal injections [74, 75]. Improvements in this issue were obtained with modified capsids [76, 77]. Because acute inflammation can favor epilepsy development and maintenance and, in turn, seizure activity favors brain inflammation [78,79,80,81], anti-inflammatory prophylaxis should be considered for people approaching gene therapy (see also below).
Another factor that may strongly impact the feasibility of future gene therapies for epilepsy is the immune response. At the moment, despite advanced technological strategies (e.g., engineered “immune stealth” capsids, immune system evasion) and significant efforts to improve the specificity, inducibility, efficiency, and broad applicability of different classes of viral vectors, we still do not adequately know the short- and long-term toxicity interactions of gene therapies with the human immune system. The prevailing host immunologic responses observed with gene therapy in the CNS are against viral vector proteins [75, 82, 83] or transgene-encoded proteins [75, 84, 85]. These reactions involve both the innate and adaptive immune systems, and can occur after both systemic or local (intra-cerebral) administration of the vector. However, no major problem in this respect has been encountered with the now broad experience of gene therapy for Leber’s congenital amaurosis and spinal muscular atrophy.
Immunological drawbacks may be associated with any gene therapy strategy. Individual pre-existing immunity may prevent vector transduction of host cells or hamper repeated administration, making the treatment ineffective. In addition, it may be hypothesized that immune reactions attacking transduced cells compromise the patient’s health. Such potential adverse immunoreactivity would worsen the symptoms in patients with epilepsy, i.e., it may directly provoke spontaneous seizures [86,87,88] or interfere with anti-epileptic drugs [89,90,91] by limiting their anti-seizure effect or amplifying their undesirable effects. To reduce the cases of unwanted immune reactions, one possibility is to measure the levels of pre-existing antibodies against the designated vector in the patients and set exclusion criteria for individuals whose values are above a certain threshold [66, 92, 93]. Unfortunately, this approach may greatly limit the potential use of future gene therapies. For example, we may have to exclude up to half of the patients because of their pre-existing immunity against AAV or herpes simplex virus proteins [94, 95]. However, immunomodulatory strategies may be adopted to suppress vector-related immune reactions. Single or combined administration of immunosuppressants has been used in many CNS-targeting gene therapies, allowing repeated administration of viral vectors and thereby prolonging the expression of transgene-encoded proteins [66, 75, 96].
7 Clinical Trials of Anti-Epileptic Gene Therapy
Over the past few decades, gene therapy has offered unique treatment opportunities for CNS diseases in which symptom management is currently the only treatment option. The recent success of the first CNS gene therapies has encouraged the growing community of researchers, and led to an escalated launch of new clinical-stage biotech companies. This sector is becoming increasingly strong, contributing to the value of the global market for all gene therapies, which is expected to reach nearly $20 billion by 2027 [97]. Its main applications are genetic diseases caused by single mutations with a loss of gene function, but also more complex progressive neurological diseases, including Alzheimer’s disease [98], Parkinson’s disease [99], and epilepsy [100].
As for epilepsy, gene therapy may first of all represent a unique opportunity for people with focal epilepsies. In fact, it may be initially offered to patients with drug-resistant focal epilepsies selected for surgical resection [101]. Gene therapy vectors may be injected directly into the epileptogenic focus, abolishing or lowering the risk of affecting healthy brain tissue and, therefore, unpredictable side effects. Should the treatment not prove effective or well tolerated, patients could undergo resective surgery as originally planned. Should it instead prove effective, patients could avoid surgery. In the wider context of future gene therapy for epilepsy, such proof of efficacy would extend the application of the treatment to some of the patients with focal epilepsies who are not eligible for surgery.
The first approved gene therapy trial is based on a randomized blinded pre-clinical study in which a non-integrating lentiviral vector was engineered to deliver a potassium channel (EKC) in excitatory neurons [43]; this EKC gene therapy was found to be effective in models of both focal neocortical and temporal lobe epilepsy, providing strong support for clinical development. Indeed, a phase I/IIa, first-in-human, open-label, single-site trial (ClinicalTrials.gov Identifier: NCT04601974) was approved in 2023 and will soon start to recruit patients with refractory neocortical epilepsy, eligible for surgical resection. Selected patients will receive a single dose of LV gene therapy treatment via an intracerebral infusion into the area scheduled for resection. The safety, tolerability, and feasibility of the treatment, including the surgical procedures, are the primary outcomes of this study. A secondary exploratory analysis will investigate delayed-onset adverse events and some efficacy indicators, such as seizure frequency and severity over a long follow-up period.
Promising therapeutic approaches that may soon lead to clinical trials for focal epilepsies have been developed using neuropeptides. The first stems from years of intensive research on the role of the inhibitory peptide NPY in the epileptic brain [50]. An AAV gene therapy product has been developed, called CG01, that over-expresses NPY and one of its receptors, Y2 [102]. This product had been licensed to Spark Therapeutics™, but will return to CombiGene in early 2024. Similarly, EpiBlok Therapeutics GmbH is developing an AAV vector overexpressing dynorphin, an opioid peptide that has been shown to induce the long-term suppression of seizures and prevention of learning and memory decline [45]. However, neither Spark Therapeutics™ or EpiBlok Therapeutics GmbH has yet announced clinical trials of these two gene therapy approaches.
Another viral vector-based gene therapy clinical trial has been recently approved for a genetic form of epilepsy, DS (ClinicalTrials.gov Identifier: NCT05419492). As described above, pre-clinical studies showed that an AAV9 vector expressing a transcription factor that increases SCN1A gene expression (ETX101) reduces spontaneous seizures and prolongs survival in a DS model [38]. Furthermore, to address the delivery system scalability, a unilateral intracerebroventricular injection of ETX101 was performed in non-human primates, and found to be well tolerated and widely distributed in the brain with low off-target transgene expression in peripheral tissues [38]. The lack of disease-modifying therapies for DS prompted the approval of the clinical trial ENDEAVOR (NCT05419492), which aims at evaluating the safety and efficacy of a one-time intracerebroventricular administration of ETX101 in infants and children with SCN1A-related DS.
8 Further Challenges
The results of these initial clinical studies will be instrumental for stimulating further research in the field, refining the current tools (in terms of target identification, regulation, and scalability) and addressing other challenges that cannot be addressed in pre-clinical settings. One such challenge, relevant for direct injections of gene therapy vectors in epileptogenic areas, is the proper coverage of the target brain tissue. This issue includes two aspects: the first is the large volume and complex geometry of the epileptogenic area in human epilepsy; the second is the need for an accurate diagnostic definition of the area that should be covered by gene therapy. With reference to the first aspect, it is known that multiple injections can be well tolerated when implementing proper neurosurgical techniques, and that the spread of the vector from the injection site can be increased by using infusion techniques such as convection-enhanced delivery. Convection-enhanced delivery has already been shown to increase the efficacy of AAV2 delivery in the brain of children [103]. With reference to the second aspect, accurate identification of the epileptogenic zone may be obtained by using imaging and electroencephalogram techniques. A promising development may be the creation of a “digital twin” of a person’s brain by combining different diagnostic data, an idea pursued by a current ongoing clinical trial (EPINOV, https://ins-amu.fr/epinov) [104]. This might help not only to better identify the origin of seizures, but also to precisely calculate the spread of gene therapy vectors needed in a specific epileptic brain.
Last, the cost of gene therapy treatments is still extremely high, very often unaffordable even for the most prosperous national healthcare systems [105]. To date, research, development and clinical trials testing gene therapy have remained restricted to high-income countries [106]. The need for sophisticated equipment, complex regulatory systems, and skilled personnel poses significant challenges to low- and middle-income countries, where the economic and social burden of epilepsy is particularly high [107]. Despite the huge health and economic potential of gene therapies, the worldwide health gap is likely to widen without concerted efforts to implement these approaches in low-resource settings.
References
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–82.
Thomas RH, Berkovic SF. The hidden genetics of epilepsy-a clinically important new paradigm. Nat Rev Neurol. 2014;10:283–92.
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–77.
Perucca E, Perucca P, White HS, Wirrell EC. Drug resistance in epilepsy. Lancet Neurol. 2023;22:723–34.
Trinka E, Rainer LJ, Granbichler CA, Zimmermann G, Leitinger M. Mortality, and life expectancy in epilepsy and status epilepticus: current trends and future aspects. Front Epidemiol. 2023;3:1081757.
Simonato M, Brooks-Kayal AR, Engel J Jr, Galanopoulou AS, Jensen FE, Moshé SL, et al. The challenge and promise of anti-epileptic therapy development in animal models. Lancet Neurol. 2014;13:949–60.
Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657–78.
West S, Nevitt SJ, Cotton J, Gandhi S, Weston J, Sudan A, et al. Surgery for epilepsy. Cochrane Database Syst Rev. 2019;6: CD010541.
Panebianco M, Rigby A, Weston J, Marson AG. Vagus nerve stimulation for partial seizures. Cochrane Database Syst Rev. 2015;2015: CD002896.
Sprengers M, Vonck K, Carrette E, Marson AG, Boon P. Deep brain and cortical stimulation for epilepsy. Cochrane Database Syst Rev. 2017;7: CD008497.
Landhuis E. The definition of gene therapy has changed. Nature. 2021. https://doi.org/10.1038/d41586-021-02736-8.
Simonato M, Bennett J, Boulis NM, Castro MG, Fink DJ, Goins WF, et al. Progress in gene therapy for neurological disorders. Nat Rev Neurol. 2013;9:277–91.
Street JS, Qiu Y, Lignani G. Are genetic therapies for epilepsy ready for the clinic? Epilepsy Curr. 2023;23:245–50.
Morris G, O’Brien D, Henshall DC. Opportunities and challenges for microRNA-targeting therapeutics for epilepsy. Trends Pharmacol Sci. 2021;42:605–16.
Choudhury SR, Hudry E, Maguire CA, Sena-Esteves M, Breakefield XO, Grandi P. Viral vectors for therapy of neurologic diseases. Neuropharmacology. 2017;120:63–80.
Ingusci S, Verlengia G, Soukupova M, Zucchini S, Simonato M. Gene therapy tools for brain diseases. Front Pharmacol. 2019;10:724.
Hudry E, Vandenberghe LH. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron. 2019;101:839–62.
Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21:255–72.
Hoy SM. Delandistrogene moxeparvovec: first approval drugs. Drugs. 2023;83(14):1323–9. https://doi.org/10.1007/s40265-023-01929-x.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–78.
Chen W, Yao S, Wan J, Tian Y, Huang L, Wang S, et al. BBB-crossing adeno-associated virus vector: an excellent gene delivery tool for CNS disease treatment. J Control Release. 2021;333:129–38.
Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. 2018;17:641–59.
Tornabene P, Trapani I. Can adeno-associated viral vectors deliver effectively large genes? Hum Gene Ther. 2020;31:47–56.
Parr-Brownlie LC, Bosch-Bouju C, Schoderboeck L, Sizemore RJ, Abraham WC, Hughes SM. Lentiviral vectors as tools to understand central nervous system biology in mammalian model organisms. Front Mol Neurosci. 2015;8:14.
Wolff JH, Mikkelsen JG. Delivering genes with human immunodeficiency virus-derived vehicles: still state-of-the-art after 25 years. J Biomed Sci. 2022;29:79.
Kido J, Sugawara K, Nakamura K. Gene therapy for lysosomal storage diseases: current clinical trial prospects. Front Genet. 2023;14:1064924.
Tokatly Latzer I, Pearl PL. Treatment of neurometabolic epilepsies: overview and recent advances. Epilepsy Behav. 2023;142: 109181.
Lundberg C, Björklund T, Carlsson T, Jakobsson J, Hantraye P, Déglon N, et al. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr Gene Ther. 2008;8:461–73.
Papayannakos C, Daniel R. Understanding lentiviral vector chromatin targeting: working to reduce insertional mutagenic potential for gene therapy. Gene Ther. 2013;20:581–8.
David RM, Doherty AT. Viral vectors: the road to reducing genotoxicity. Toxicol Sci. 2017;155:315–25.
Asaad W, Volos P, Maksimov D, Khavina E, Deviatkin A, Mityaeva O, et al. AAV genome modification for efficient AAV production. Heliyon. 2023;9: e15071.
Penaud-Budloo M, François A, Clément N, Ayuso E. Pharmacology of recombinant adeno-associated virus production. Mol Ther Methods Clin Dev. 2018;8:166–80.
Wright JF. Product-related impurities in clinical-grade recombinant AAV vectors: characterization and risk assessment. Biomedicines. 2014;2:80–97.
Kang L, Jin S, Wang J, Lv Z, Xin C, Tan C, et al. AAV vectors applied to the treatment of CNS disorders: clinical status and challenges. J Control Release. 2023;355:458–73.
Grieger JC, Soltys SM, Samulski RJ. Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol Ther. 2016;24:287–97.
Kantor B, McCown T, Leone P, Gray SJ. Clinical applications involving CNS gene transfer. Adv Genet. 2014;87:71–124.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–22.
Tanenhaus A, Stowe T, Young A, McLaughlin J, Aeran R, Lin IW, et al. Cell-selective adeno-associated virus-mediated gene regulation therapy rescues mortality and seizure phenotypes in a Dravet syndrome mouse model and is well tolerated in nonhuman primates. Hum Gene Ther. 2022;33:579–97.
Lopez-Santiago L, Isom LL. Dravet syndrome: a developmental and epileptic encephalopathy. Epilepsy Curr. 2019;19(1):51–3.
Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure. 2017;44:58–64.
Ingusci S, Cattaneo S, Verlengia G, Zucchini S, Simonato M. A matter of genes: the hurdles of gene therapy for epilepsy. Epilepsy Curr. 2019;19:38–43.
Jensen TL, Gøtzsche CR, Woldbye DPD. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021;14: 695937.
Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, et al. Epilepsy gene therapy using an engineered potassium channel. J Neurosci. 2019;39:3159–69.
Richichi C, Lin E-JD, Stefanin D, Colella D, Ravizza T, Grignaschi G, et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24:3051–9.
Agostinho AS, Mietzsch M, Zangrandi L, Kmiec I, Mutti A, Kraus L, et al. Dynorphin-based “release on demand” gene therapy for drug-resistant temporal lobe epilepsy. EMBO Mol Med. 2019;11: e9963.
Paradiso B, Marconi P, Zucchini S, Berto E, Binaschi A, Bozac A, et al. Localized delivery of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model. Proc Natl Acad Sci USA. 2009;106:7191–6.
Woldbye DPD, Angehagen M, Gøtzsche CR, Elbrønd-Bek H, Sørensen AT, Christiansen SH, et al. Adeno-associated viral vector-induced overexpression of neuropeptide Y Y2 receptors in the hippocampus suppresses seizures. Brain. 2010;133:2778–88.
Kätzel D, Nicholson E, Schorge S, Walker MC, Kullmann DM. Chemical-genetic attenuation of focal neocortical seizures. Nat Commun. 2014;5:3847.
Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med. 2012;4:152–61.
Cattaneo S, Verlengia G, Marino P, Simonato M, Bettegazzi B. NPY and gene therapy for epilepsy: how, when,... and Y. Front Mol Neurosci. 2020;13: 608001.
Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, et al. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol Ther. 2020;28:235–53.
Colasante G, Qiu Y, Massimino L, Di Berardino C, Cornford JH, Snowball A, et al. In vivo CRISPRa decreases seizures and rescues cognitive deficits in a rodent model of epilepsy. Brain. 2020;143:891–905.
Geisler A, Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J Exp Med. 2016;6:37–54.
Lisowski L, Tay SS, Alexander IE. Adeno-associated virus serotypes for gene therapeutics. Curr Opin Pharmacol. 2015;24:59–67.
Qiu Y, O’Neill N, Maffei B, Zourray C, Almacellas-Barbanoj A, Carpenter JC, et al. On-demand cell-autonomous gene therapy for brain circuit disorders. Science. 2022;378:523–32.
Jazwa A, Florczyk U, Jozkowicz A, Dulak J. Gene therapy on demand: site specific regulation of gene therapy. Gene. 2013;525:229–38.
Rhim T, Lee DY, Lee M. Hypoxia as a target for tissue specific gene therapy. J Control Release. 2013;172:484–94.
Bochkov YA, Palmenberg AC. Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques. 2006;41:283–4 (288 passim).
Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, et al. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep. 2017;7:2193.
Hadpech S, Jinathep W, Saoin S, Thongkum W, Chupradit K, Yasamut U, et al. Impairment of a membrane-targeting protein translated from a downstream gene of a “self-cleaving” T2A peptide conjunction. Protein Expr Purif. 2018;150:17–25.
Melin E, Andersson M, Gøtzsche CR, Wickham J, Huang Y, Szczygiel JA, et al. Combinatorial gene therapy for epilepsy: gene sequence positioning and AAV serotype influence expression and inhibitory effect on seizures. Gene Ther. 2023. https://doi.org/10.1038/s41434-023-00399-w.
Nathwani AC, Tuddenham EGD, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–65.
Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–86.
Sinnett SE, Gray SJ. Recent endeavors in MECP2 gene transfer for gene therapy of Rett syndrome. Discov Med. 2017;24:153–9.
Gadalla KKE, Vudhironarit T, Hector RD, Sinnett S, Bahey NG, Bailey MES, et al. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol Ther Methods Clin Dev. 2017;5:180–90.
Shen W, Liu S, Ou L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: a meta-analysis. Front Immunol. 2022;13:1001263.
Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369:2097–105.
Muramatsu S-I, Fujimoto K-I, Kato S, Mizukami H, Asari S, Ikeguchi K, et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther. 2010;18:1731–5.
Palfi S, Gurruchaga JM, Ralph GS, Lepetit H, Lavisse S, Buttery PC, et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase ½ trial. Lancet. 2014;383(9923):1138–46.
Palfi S, Gurruchaga JM, Lepetit H, Howard K, Ralph GS, Mason S, et al. Long-term follow-up of a phase I/II study of ProSavin, a lentiviral vector gene therapy for Parkinson’s disease. Hum Gene Ther Clin Dev. 2018;29(3):148–55.
Hordeaux J, Hinderer C, Goode T, Buza EL, Bell P, Calcedo R, et al. Toxicology study of intra-cisterna magna adeno-associated virus 9 expressing iduronate-2-sulfatase in rhesus macaques. Mol Ther Methods Clin Dev. 2018;14(10):68–78.
Hordeaux J, Hinderer C, Goode T, Katz N, Buza EL, Bell P, et al. Toxicology study of intra-cisterna magna adeno-associated virus 9 expressing human alpha-L-iduronidase in rhesus macaques. Mol Ther Methods Clin Dev. 2018;14(10):79–88.
Flotte TR. Revisiting the “new” inflammatory toxicities of adeno-associated virus vectors. Hum Gene Ther. 2020;31(7–8):398–9.
Perez BA, Shutterly A, Chan YK, Byrne BJ, Corti M. Management of neuroinflammatory responses to AAV-mediated gene therapies for neurodegenerative diseases. Brain Sci. 2020. https://doi.org/10.3390/brainsci10020119.
Prasad S, Dimmock DP, Greenberg B, Walia JS, Sadhu C, Tavakkoli F, et al. Immune responses and immunosuppressive strategies for adeno-associated virus-based gene therapy for treatment of central nervous system disorders: current knowledge and approaches. Hum Gene Ther. 2022;33:1228–45.
Sen D. Improving clinical efficacy of adeno associated vectors by rational capsid bioengineering. J Biomed Sci. 2014;21:103.
Lugin ML, Lee RT, Kwon YJ. Synthetically engineered adeno-associated virus for efficient, safe, and versatile gene therapy applications. ACS Nano. 2020;14:14262–83.
Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40.
Sankar R, Auvin S, Mazarati A, Shin D. Inflammation contributes to seizure-induced hippocampal injury in the neonatal rat brain. Acta Neurol Scand Suppl. 2007;186:16–20.
Pracucci E, Pillai V, Lamers D, Parra R, Landi S. Neuroinflammation: a signature or a cause of epilepsy? Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22136981.
Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. 2019;15:459–72.
Bessis N, GarciaCozar FJ, Boissier M-C. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11(Suppl. 1):S10–7.
Thaci B, Ulasov IV, Wainwright DA, Lesniak MS. The challenge for gene therapy: innate immune response to adenoviruses. Oncotarget. 2011;2:113–21.
Broberg EK, Hukkanen V. Immune response to herpes simplex virus and gamma134.5 deleted HSV vectors. Curr Gene Ther. 2005;5:523–30.
Gougeon M-L, Poirier-Beaudouin B, Ausseil J, Zérah M, Artaud C, Heard J-M, et al. Cell-mediated immunity to NAGLU transgene following intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome. Front Immunol. 2021;12: 655478.
Yamaguchi H, Tanaka T, Maruyama A, Nagase H. Septic encephalopathy characterized by acute encephalopathy with biphasic seizures and late reduced diffusion and early nonconvulsive status epilepticus. Case Rep Neurol Med. 2016;2016:7528238.
Semple BD, Dill LK, O’Brien TJ. Immune challenges and seizures: how do early life insults influence epileptogenesis? Front Pharmacol. 2020;11:2.
Ahl M, Taylor MK, Avdic U, Lundin A, Andersson M, Amandusson Å, et al. Immune response in blood before and after epileptic and psychogenic non-epileptic seizures. Heliyon. 2023;9: e13938.
Beghi E, Shorvon S. Antiepileptic drugs and the immune system. Epilepsia. 2011;52(Suppl. 3):40–4.
Mullan KA, Anderson A, Illing PT, Kwan P, Purcell AW, Mifsud NA. HLA-associated antiepileptic drug-induced cutaneous adverse reactions. Hladnikia. 2019;93:417–35.
De Ponti F, Lecchini S, Cosentino M, Castelletti CM, Malesci A, Frigo GM. Immunological adverse effects of anticonvulsants: what is their clinical relevance? Drug Saf. 1993;8:235–50.
Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther. 2020;28:723–46.
Perocheau DP, Cunningham S, Lee J, Antinao Diaz J, Waddington SN, Gilmour K, et al. Age-related seroprevalence of antibodies against AAV-LK03 in a UK population cohort. Hum Gene Ther. 2019;30:79–87.
Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–12.
Herrlinger U, Kramm CM, Aboody-Guterman KS, Silver JS, Ikeda K, Johnston KM, et al. Pre-existing herpes simplex virus 1 (HSV-1) immunity decreases, but does not abolish, gene transfer to experimental brain tumors by a HSV-1 vector. Gene Ther. 1998;5:809–19.
Au HKE, Isalan M, Mielcarek M. Gene therapy advances: a meta-analysis of AAV usage in clinical settings. Front Med. 2021;8: 809118.
Market Data Forecast. Gene therapy market size, share, trends & growth, 2022 to 2027. January 2022. Available from: https://www.marketdataforecast.com/market-reports/gene-therapy-market. (Accessed 7 Dec 2023).
Sudhakar V, Richardson RM. Gene therapy for neurodegenerative diseases. Neurotherapeutics. 2019;16:166–75.
Axelsen TM, Woldbye DPD. Gene therapy for Parkinson’s disease, an update. J Parkinsons Dis. 2018;8:195–215.
Drew L. Gene therapy targets epilepsy. Nature. 2018;564:S10–1.
Jobst BC, Cascino GD. Resective epilepsy surgery for drug-resistant focal epilepsy: a review. JAMA. 2015;313:285–93.
Szczygieł JA, Danielsen KI, Melin E, Rosenkranz SH, Pankratova S, Ericsson A, et al. Gene therapy vector encoding neuropeptide Y and its receptor Y2 for future treatment of epilepsy: preclinical data in rats. Front Mol Neurosci. 2020;13:232.
Pearson TS, Gupta N, San Sebastian W, Imamura-Ching J, Viehoever A, Grijalvo-Perez A, et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat Commun. 2021;12:4251.
Naddaf M. How virtual models of the brain could transform epilepsy surgery. Nature. 2023;616:227–8.
Gene therapies should be for all. Nat Med. 2021;27:1311. https://doi.org/10.1038/s41591-021-01481-9.
World Economic Forum. Accelerating global access to gene therapies: case studies from low- and middle-income countries. Available from: https://www.weforum.org/whitepapers/accelerating-global-access-to-gene-therapies-case-studies-from-low-and-middle-income-countries/. (Accessed 18 Oct 2022).
Beghi E. The epidemiology of epilepsy. Neuroepidemiology. 2020;54:185–91.
Author information
Authors and Affiliations
Contributions
BB and SC introduced on epilepsies and viral vectors and discussed strategies for optimizing gene therapy approaches; SZ focused on clinical trials of antiepileptic gene therapy; MS and MaS supervised and reviewed the opinion article.
Corresponding author
Ethics declarations
Funding
This work was supported by funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 964712 (Prime).
Conflict of Interest
Barbara Bettegazzi, Stefano Cattaneo, Michele Simonato, Silvia Zucchini, and Marie Soukupova have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bettegazzi, B., Cattaneo, S., Simonato, M. et al. Viral Vector-Based Gene Therapy for Epilepsy: What Does the Future Hold?. Mol Diagn Ther 28, 5–13 (2024). https://doi.org/10.1007/s40291-023-00687-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00687-6