
Abstract
Multiple sclerosis (MS) is a chronic autoimmune demyelinating and neurodegenerative disease of the central nervous system with a wide variety of clinical phenotypes. In spite of the phenotypic classification of MS patients, current data provide evidence that diffuse neuroinflammation and neurodegeneration coexist in all MS forms, the latter gaining increasing clinical relevance in progressive phases. Given that the transition phase of relapsing-remitting MS (RRMS) to secondary progressive MS (SPMS) is not well defined, and widely accepted criteria for SPMS are lacking, randomised controlled trials (RCTs) specifically designed for the transition phase have not been conducted. This review summarizes primary and secondary analyses and reports derived from phase III prospective clinical RCTs listed in PubMed of compounds authorised through the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) for the treatment of MS. The best data are available for interferon beta-1a (IFNb-1a) subcutaneous (s.c.), IFNb-1b s.c., mitoxantrone and siponimod, the latter being the most modern compound with likely the best risk-to-effect ratio. Moreover, there is a labels discrepancy for many disease-modifying treatments (DMTs) between the FDA and EMA, which have to be taken into consideration when opting for a specific DMT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The transition phase of relapsing-remitting MS (RRMS) to secondary progressive MS (SPMS) is not well defined. Clinical trials have thus not been conducted in these patients. |
None of the currently available disease-modifying treatments is specifically approved for this phenotype. |
Potential efficacy may be deduced from pivotal phase III trials and respective post hoc analysis. |
The best data are available for siponimod, interferon beta-1a subcutaneous, and historically mitoxantrone. |
1 Clinical Phenotypes of Multiple Sclerosis (MS)
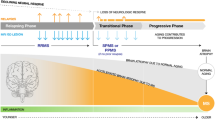
Multiple sclerosis (MS) is the most prevalent chronic inflammatory, degenerative and demyelinating disease of the central nervous system (CNS), and manifests itself with a wide spectrum of clinical phenotypes. According to the classification proposal of Lublin et al. [1], the core MS phenotypes are relapsing-remitting (RR) disease along with progressive disease (PMS). Relapsing remitting multiple sclerosis (RRMS) is characterised by a clinical course with defined recurrent attacks of new or exacerbated neurological dysfunction. Clinically isolated syndrome (CIS) is defined as the first episode of neurological symptoms caused by inflammation or demyelination and is now considered as a component of the RR phenotype provided that the criteria of dissemination in time are fulfilled [2]. PMS consists of two subtypes: primary progressive (PPMS) and secondary progressive (SPMS). SPMS is marked by gradual accumulation of disability following an initially RR course of disease, whereas PPMS is characterized by insidious progression without relapses. Despite the phenotypic categorization of MS, these forms share common modifiers of the disease status. Focal inflammatory disease activity distinguished by clinical relapses and magnetic resonance imaging (MRI) criteria (gadolinium-enhancing lesions or new or enlarging T2 lesions) and disease progression deriving from clinical assessment are useful descriptors for both relapsing-remitting and progressive forms. In this context, current data suggest that neurodegeneration and diffuse neuroinflammation already occurs early on in RRMS as well, and that MS may be observed as a spectrum of coexisting neuroinflammation and neurodegeneration in spite of the terminology [3]. A recent publication spearheaded the concept of progressive disability accumulation occurring independently of relapse activity [4]. This proportion of disability is likely to be driven by a smouldering pathological process that may affect the entire CNS [4].
2 Progression Independent of Relapse Activity (PIRA)
The arising question is whether all patients with MS at some point present signs of a progressive course. A new term that was recently introduced is progression independent of relapse activity (PIRA), emerging from a data-analysis of the Tysabri Observational Program (TOP), a prospective open-label study in RRMS patients receiving intravenous natalizumab for a median of 108.3 weeks [5]. It refers to the amount of accumulated neurological disability occurring independent of relapse activity, a feature that characterizes PMS that is believed to be linked to neurodegeneration and/or diffuse inflammatory processes involving innate immunity. The Swiss Multiple Sclerosis Cohort study (n = 1335) used a roving Expanded Disability Status Scale (EDSS) to evaluate MS patients over a median of 4 years of follow-up [6]. A relevant proportion of patients (15%) with CIS/RRMS experienced PIRA within 6 years, and PIRA accounted for 62% of confirmed disability progression (CDP) events in these patients. In the SPMS/PPMS group, 93–95% of CDP events were attributed to PIRA. Another Swiss PIRA-analysis (n = 1640) aimed to compare fingolimod with platform injectables [7]. Overall, PIRA was observed in 3.1% of the patients under treatment with interferon-beta (IFNb)/glatiramer acetate and 4.1% of the patients on fingolimod. CDP was observed in 137 patients (8.4%): 92 patients (8.8%) in the IFNb/glatiramer acetate and 45 (7.6%) in the fingolimod group, of which 32 (34.8%) and 24 (53.3%) were PIRA, respectively. A retrospective, cross-sectional study of clinical data from two German MS tertiary referral centres demonstrated that patients who are initiated on natalizumab early during disease course, usually in order to treat an aggressive clinical phenotype, have a higher risk of early PIRA probably as a result of an indication bias [8]. Recent clinical study data further suggest that PIRA already starts in early RRMS and becomes the main driver of disability accumulation as the disease progresses [9, 10]. In the pooled analysis of the two OPERA trials both the IFNb-1a and ocrelizumab groups demonstrated a high proportion of 24-week confirmed worsening or progression (CDP) after 96 weeks (78 and 87%, respectively), which was associated with neurological worsening independent of overt relapses [11].
The above-mentioned study data provide clinical evidence of an underlying progressive course in all MS patients independently of the disease classification. However, as the PIRA concept is only based on clinical criteria, disability accrual may also result from focal inflammatory activity associated with MRI lesions. Therefore, the EMA guideline for SPMS suggests that in order to evaluate the efficacy of a product against disability progression in SPMS, it is recommended to target only SPMS patients without a recent relapse and no MRI activity suggestive of active inflammation, and with evidence of recent progression independent of relapses [12].
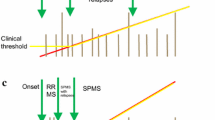
3 Defining the Transition Phase to Secondary Progressive MS (SPMS)
The majority of MS patients (∼ 85%) are diagnosed with RRMS, and approximately 50–60% of them transition to SPMS between 5 and 30 years [13, 14], highlighting the heterogeneity of the time between disease onset and transition to SPMS due to the high interindividual variability in the disease course. According to the revised Lublin criteria, SPMS is diagnosed retrospectively based on a course of confirmed progression over the last 3–12 months with or without acute exacerbations during the progressive course after an initial relapsing disease course [1]. The high variability in the disease course, the overlap in pathophysiological mechanisms between relapsing and progressive MS [3, 15, 16], together with clinical difficulties in the detection of progression, render the identification of transition of RRMS to SPMS a major diagnostic challenge. To date, consented criteria defining SPMS do not exist. Based on data analysis from a multinational MS registry [17], Lorscheider et al. proposed an objective definition of SPMS with the best performance from a pool of 576 candidate definitions. This definition consists of:
-
(1)
A disability progression by 1 EDSS step in patients with EDSS < 6 or 0.5 EDSS steps in patients with EDSS ≥ 6 in the absence of a relapse;
-
(2)
A minimum EDSS score of 4 including a pyramidal functional system (FS) score of at least 2; and
-
(3)
Confirmed progression over 3 months, including confirmation within the leading FS [18].
In the clinical study of Kopp et al. [19], slightly modified inclusion criteria from the EXPAND trial were applied on the Danish nationwide MS population with a diagnosis of clinical SPMS assigned by an MS-neurologist and RRMS patients fulfilling the MSBase diagnostic definition for conversion to SPMS. The m-EXPAND criteria identify patients with recent worsening on the EDSS score likely not explained by a recent relapse:
-
(1)
An EDSS from 3.0 to 6.5 (both inclusive) (at index date +/− 6 months); and
-
(2)
EDSS progression within the last 2 years before data extraction, defined as EDSS progression of 1 point or more in patients with an EDSS score of less than 6.0 or ≥ 0.5 point in patients with EDSS score ≥ 6.0, in the absence of relapses 6 months prior to progression and EDSS ≥ 3.0 at time of progression.
-
(3)
Disability progression as described above confirmed over ≥ 6 months.
The MSBase SPMS definition captured ~ 20% of Danish RRMS patients at putative high risk of converting to SPMS or who may already have converted to SPMS.
From a regulatory perspective, different proposals for SPMS definitions may impair the comparability of results from different trials investigating the effect of medical products on SPMS.
Due to the aforementioned factors, a consequent diagnostic uncertainty, which translates into a significant delay in the diagnosis of SPMS, is observed [18, 20, 21]. According to a retrospective cohort study, which reviewed 123 MS patients with a long-term clinical follow-up of ≥ 8 years, described a period of diagnostic uncertainty regarding the transition from RRMS to SPMS in a significant patient proportion with a mean duration of 2.9 ± 0.8 years [21]. A further retrospective multicentre cohort study in Argentina (n = 170) aiming to describe the length of time required to reclassify RRMS patients who have clinically transitioned to SPMS demonstrated a period of diagnostic uncertainty regarding the transition from RRMS to SPMS of 3.3 years [22]. Although progression is driven mostly through neurodegenerative changes, inflammation-associated neuroaxonal loss along with new or enlarging T2/FLAIR lesions in the MRI are also present in progressive MS forms [23, 24].
Furthermore, the overlap in pathophysiological mechanisms between relapsing and progressive MS [3, 15, 16] hampers the identification and validation of specific and sensitive imaging and/or biological markers for monitoring progression and identifying the transition of RRMS to SPMS. The associations of serum neurofilament light chain (sNfL) with current or future disability appear to be inconsistent, as recently reviewed [25]. Multiple studies have demonstrated a significant association between the disease progression of MS patients and evoked potentials [26,27,28,29] or peripapillary retinal nerve fibre layer (pRNFL) thickness assessed by optical coherence tomography (OCT) [30,31,32]. Additionally, MRI cerebral and spinal cord atrophy have been correlated with neurodegeneration in progressive MS [33, 34].
Further, due to the gradual progression of disease, there is an overlap of symptoms and MRI features between RRMS and SPMS that renders the clinical detection of the onset of progression difficult. Therefore, it often remains unnoticed by patients as wells as physicians. Furthermore, recent clinical study data suggest that insidious disability attributable to silent progression may be present in the early stages of MS [35, 36]. The EDSS has long been viewed as the gold-standard tool for evaluation of disability and clinical disease progression in MS [37].
However, EDSS focuses on ambulatory disability in the middle and upper end of the scale (scores >3.0), and is less sensitive to other aspects of impairment in MS such as cognitive function. Cognitive impairment can emerge at different stages of MS. It is associated with a worse prognosis in the early RRMS disease phases and CIS patients with cognitive impairment are at higher risk to develop clinically defined MS [38, 39]. Therefore, cognitive assessment using a screening test, such as symbol digit modalities test (SDMT), can be of great importance in the clinical follow-up of MS patients and the early detection of disease progression. However, the SDMT can be considered a useful test to evaluate mental processing speed but not a validated measure for cognitive function [40]. A further quantitative instrument that enables measurement of arm/hand dexterity and cognitive function in MS patients is the Multiple Sclerosis Functional Composite (MSFC) [41]. In more recent studies, composite scores integrating several established tests like EDSS, timed 25-foot walk (T25FW), SDMT and 9-hole peg test (9HPT) have been increasingly implemented as clinical outcome measures to increase the sensitivity for disability change [11, 42]. However, concerning the design of randomised controlled trials (RCTs) focussed on the transition phase, SMDT, T25FW and 9HPT have not been established as primary endpoints but as secondary endpoints. Further, T25FW and 9HPT could be included in a composite primary endpoint combined with EDSS, and have to be correlated to the clinical relevance of the observed effects. For this reason, it is recommended to include additional functional endpoints in order to relate the effect size of composite scores to a clinical relevance (e.g., patient reported outcomes) [40]. The MSProDiscuss is an additional exploratory clinical tool designed to detect signs of secondary progressive disease through a structured interaction between physicians and patients raising awareness of the risk of transition from RRMS to SPMS [43].
Taken together, current data provide evidence that PIRA is a significant indicative component of the transition phase of MS. However, recent clinical study findings indicate that insidious disability progression appears even in the earliest phases of the disease [9, 10]. Therefore, PIRA represents a negative prognostic factor for further disability accumulation in the disease course.
4 Treatment Options for the Transition Phase of MS
Given that the transition phase is not well defined and widely accepted criteria for SPMS are lacking, RCTs specifically designed for the transition phase have not been conducted. The compounds that met their primary endpoints in RCTs for this specific condition are IFNb, i.e. IFNb-1a subcutaneous (s.c). and IFNb-1b s.c., the S1P receptor modulator siponimod, and mitoxantrone (Fig. 1). IFNb-1a s.c. underwent two phase III RCTs, PRISMS [44] and SPECTRIMS [45]; while PRISMS only enrolled RRMS patients, SPECTRIMS focussed on SPMS patients. While PRISMS met its primary endpoint [44], SPECTRIMS did not [45]. However, in a recent post hoc analysis, Freedman et al. identified an effect on clinical and MRI parameters in those patients deemed to transition between RRMS and SPMS [46]. IFNb-1b s.c. has also been evaluated in RRMS patients as well as in SPMS patients [47]. However, only the European SPMS trial met its primary endpoint of confirmed disability progression [48]. There was no significant difference for this primary endpoint in a similar trial conducted by the North American Study Group when compared to placebo-treated SPMS patients [49]. A post hoc analysis adressing the putative transition phase has not been conducted. Consequently, IFNb-1a s.c. is licensed for RRMS and SPMS under the US Food and Drug Administration (FDA) label, and for RMS under the European Medicines Agency (EMA) label, with a specific mention that efficacy has not been demonstrated in patients with SPMS without ongoing relapse activity. In contrast, IFNb-1b s.c. was granted approval for the treatment of CIS, RRMS and active SPMS evidenced by relapse activity by the EMA and for the treatment of RMS by the FDA (Table 1).

Overview of disease-modifying agents for multiple sclerosis (MS). Flag of the European Union: authorisation through EMA, Flag of the United States of America: authorisation through FDA. *Not authorised under centralized EMA procedure. Indication granted by decentralized approval. ALEM alemtuzumab, CIS clinically isolated syndrome, CLAD cladribine, DMF dimethyl fumarate, DRF diroximel fumarate, EMA European Medicines Agency, FDA US Food and Drug Administration, FTY fingolimod, GLAT glatiramer acetate, IFNb-1a interferon beta-1a, IFNb-1b interferon beta-1b, MITOX mitoxantrone, MMF monomethyl fumarate, NAT natalizumab, OCR ocrelizumab, OFA ofatumumab, OZA ozanimod, PbO placebo, peg IFNb-1a pegylated interferon beta-1a, PON ponesimod, PPMS primary progressive multiple sclerosis, RIS radiologically isolated syndrome, RMS relapsing multiple sclerosis, RRMS relapsing-remitting multiple sclerosis, SIPO siponimod, SPMS secondary progressive multiple sclerosis (+R with relapses, −R without relapses), TER teriflunomide
Siponimod’s safety and efficacy were initially addressed in RRMS patients in the phase II study BOLD [50, 51]. However, the phase III RCT EXPAND only included SPMS patients, both with and without superimposed relapses. While BOLD was not designed and thus underpowered to detect an effect on annualized relapse rate reduction or EDSS progression, EXPAND met its primary endpoint, i.e. 3-month confirmed disability progression. At the time of the evaluation, the indication applied was the treatment of SPMS patients. However, from the EMA’s perspective, it was challenging to disentangle the effect of siponimod on disability progression driven by the effect on relapses, and based on provided results, the effect of siponimod on disability progression was judged to be small in patients without relapses and without focal MRI activity. Consequently, this has resulted in an EMA approval for active SPMS as defined by the Lublin criteria [1], rather than the entire SPMS spectrum [12]. In contrast, the FDA granted approval for RMS, which includes CIS, RRMS as well as SPMS with ongoing disease activity. Patients transitioning from relapsing to progressive MS may, thus, effectively be treated with siponimod under both the EMA and the FDA labels.
Another highly active DMT with positive phase III data in a cohort of MS patients specifically including SPMS is mitoxantrone, which was tested in the MIMS trial involving both progessing relapsing MS but also SPMS patients [52]. In this study, mitoxantrone demonstrated substantial effects on disability progression, which was reduced by more than half as compared to with placebo (8 vs. 22%). Mitoxantrone has been approved as a generic medication for SPMS, progressive RMS and worsening RRMS by the FDA and for highly active RMS by the EMA. The less favourable safety profile in comparison to other MS DMTs, namely the cardiac side effects and secondary malignancies, have increasingly limited the use of mitoxantrone in Europe and it is hardly ever used in the USA.
In spite of minute proportions of SPMS patients enrolled in RCTs and thus a paucity of high-level scientific evidence gathered specifially on SPMS patients, a number of compounds have been approved for the treatment of RMS patients including active SPMS. Under the assumption that relapses in RRMS and SPMS are likely to have the same underlying inflammatory pathophysiology, it is reasonably justified to extrapolate efficacy on relapses in RRMS patients to the efficacy on relapses in SPMS, even though the proportions of SPMS patients were small in phase III RCTs focused on RMS. This extrapolation, however, cannot be considered appropriate for the effects on disability accumulation as pathophysiology is different in RRMS and PMS [12].
Ocrelizumab was successfully tested in RMS (OPERA I and II) as well as PPMS (ORATORIO) [53, 54]. The endpoints of reducing the risk for 3-month CDP and 6-month CDP were met in OPERA I/II as well as in ORATORIO. In a post hoc efficacy analysis, disability outcomes were assessed in those patients who had a baseline EDSS ≥ 4.0 [55]. From OPERA I and II 375 RMS patients were included, and n = 507 PPMS patients from the ORATORIO trial. These patients were on average 40.2–44.9 years of age with a mean time since MS symptom onset of 6.73–10.25 years. There was a significant relative risk reduction in 3-month and 6-month CDP for ocrelizumab-treated versus IFNb-1a-treated RMS patients [56]. This patient cohort, i.e. age > 40 years and EDSS > 3.5, arguably resembles the conditions of patients transitioning from RRMS to SPMS, utilized by Freedman et al. [46]. Ocrelizumab was not specifically investigated in SPMS. In the ORATORIO PPMS trial a hazard ratio (HR) = 0.71 in favour of ocrelizumab-treated PPMS patients compared to placebo-treated patients was observed [55].
Ofatumumab was successfully tested over teriflunomide in RRMS as well as SPMS (ASCLEPIOS I and II) [57]. The endpoints of reducing the risk for 3-month CDP and 6-month CDP were met in ASCLEPIOS I/II. In a post hoc efficacy analysis, ofatumumab treatment of RMS patients older than 40 years or affected by a baseline EDSS > 3.5 was effective with regards to annualized relapse rate reduction [58].
The Clarity phase III RCT testing cladribine versus placebo enrolled participants with RRMS only [59]. Clarity met its primary endpoint, i.e., ARR reduction, as well as the secondary endpoint of 3-month CDP risk reduction. In addition, in a post hoc subgroup analysis cladribine treatment of RRMS patients older than 40 years or affected by a baseline EDSS ≥ 3.5 was effective with regards to relapse rate reduction [60]. RCTs testing cladribine in progressive MS have not been conducted.
For fumarate treatment, i.e., first-generation dimethyl fumarate and second-generation diroximel fumarate, the FDA granted a broad RMS label. In contrast, the EMA label for dimethyl fumarate is RRMS, and considering that the evidence on efficacy for diroximel fumarate fully relies on the evidence from pivotal trials for dimethyl fumarate, the same label was applied and granted for diroximel fumarate. In fact, DEFINE and CONFIRM enrolled RRMS patients only. DEFINE, but not CONFIRM, met its secondary endpoint of CDP risk reduction over placebo treatment [61, 62]. In a post hoc analysis, dimethyl fumarate treatment of RRMS patients older than 40 years or affected by a baseline EDSS ≥ 2.0 was effective with regards to relapse rate reduction [63]. An additional analysis was performed for 3-month CDP; however, dimethyl fumarate treatment was not favourable over placebo treatment for either patients > 40 years of age or with a baseline EDSS ≥ 2.0 [63].
The TEMSO and TOWER phase III RCTs demonstrated that teriflunomide significantly reduced the relapse rate and MRI activity in RMS patients compared to placebo, while effects on CDP were significant in the TEMSO study with teriflunomide at 14 mg as well as in the TOWER study [64]. The TENERE phase III RCT reported better adherence and tolerability of teriflunomide compared to IFNb-1a in RMS patients, while no significant differences in relapse rate reduction were observed [90]. A post hoc analysis of the TEMSO data revealed that teriflunomide versus placebo effects on the ARR were consistent in patients older than 38 years, but lost significance for patients classified as SPMS and for patients with EDSS > 3.5 at baseline. Notably, only 60 patients with SPMS were analysed. There was no significantly different effect revealed for 3-month CDP in any of these subgroups [65]. Teriflunomide was not specifically investigated in SPMS.
In contrast to siponimod, the other S1P modulators fingolimod, ozanimod and ponesimod have not been evaluated in trials including only a SPMS population. All have demonstrated efficacy on relapse rates and have been licensed for the treatment of RMS by the FDA and for the treatment of RRMS by EMA, except for ponesimod for which the indication included RMS with active disease as applied by the applicant. Significant effects on CDP were only observed in comparison to placebo and only in one of the two FREEDOMS trials, while FREEDOMS II and the active comparator controlled trials of fingolimod, ozanimod and ponesimod failed to reach significance regarding this outcome. The largest body of post hoc analyses of phase III data for different subgroups is available for fingolimod and revealed that the effects on annulised relapse rate (ARR) reduction are retained in patients older than 40 years, those with a baseline EDSS > 3.0, and those with a disease duration of at least 3 years [66]. Fingolimod was additionally investigated in PPMS; however, the INFORMS RCT failed to meet its primary endpoint of 3-month CDP in patients treated for at least 3 years with fingolimod [67]. In a recent subgroup analysis of the OPTIMUM trial, ponesimod was shown to be superior in early MS patients, as defined as those with EDSS < 3.5 or DMT-naive patients; the treatment effect in those patients with EDSS of 3.5 or above was below the entire ponesimod group [68].
Natalizumab is arguably one of the most effective DMTs in reducing relapse rates and has also demonstrated strong effects on CDP in RMS patients, which was documented in the AFFIRM trial [69]. At the same time, it is one of the few highly effective MS DMTs, which was additionally investigated in a designated phase III SPMS RCT, ASCEND [70]. However, the ASCEND trial did not meet its primary outcome of reducing confirmed EDSS progression and was also negative for the T25FW. Conversly, it did reveal positive effects on disability progression of the upper limb assessed by the 9HPT. In a subgroup analysis of the AFFIRM trial when looking at ARR, natalizumab was significantly more effective in patients older than 40 years compared to placebo, but not in patients with a baseline EDSS > 3.5 [71]. A subgroup analysis was also conducted concering effects on CDP; however, neither patients older than 40 years nor those with a baseline EDSS > 3.5 benefited more than from placebo treatment [71].
Alemtuzumab has been investigated in cohorts of RRMS patients in the phase III RCTs CARE-MS I and II [72, 73], demonstrating significant effects on ARR and MRI measures compared to IFNb1a but significant effects on CDP only in CARE-MS II, which included patients with insufficient response to prior therapies. Notably, CARE-MS I only enrolled DMT-naïve patients with a baseline EDSS ≤ 3.0, resembling early RRMS patients [72]. Both the FDA and the EMA have approved alemtuzumab for RRMS and recommend the use only for patients with insufficient response to other DMTs as a result of safety concerns linked to autoimmunity. A formal subgroup analysis of the CARE-MS study populations has not been conducted. However, a post hoc analysis of the clinically centered 3-year, rater-blinded phase II study CAMMS223 showed treatment effects for patients older than 31 years and with a baseline EDSS > 2.0 with respect to the ARR and 6-month CDP [74]. In an open-label extension of both CARE-MS trials, 80% of patients remained free of CDP and 40% showed confirmed disability improvement during 6 years of observation [75]. A post hoc analysis of 811 patients included in the CARE-MS trials revealed that, at 6.2 years of follow-up, only 20 converted to SPMS (Kaplan–Meier estimate, 2.7%; 95% confidence interval (CI), 1.8–4.2). [76] However, alemtuzumab was not investigated in SPMS.
Glatiramer acetate was issued an RMS label from the FDA and the European regulatory authorities based on the results of a phase III RCT demonstrating significant effects on ARR reduction; disability progression, i.e., 3-month CDP, was not significantly reduced compared to placebo [77]. In a small post hoc analysis patients with a baseline EDSS > 3.5 did not benefit from glatiramer acetate treatment with respect to halting disease progression [78]. Notably, this subgroup consisted of only 52 patients. Glatiramer acetate was not investigated in SPMS.
5 Differences Between European Medicines Agency (EMA) and US Food and Drug Administration (FDA) Labels
Both the FDA and the EMA base the approval of new DMTs on the seminal phase III RCT data submitted. However, the final label granted for the different DMTs does not always reflect the profile of the cohorts enrolled in these phase III trials. A particular difference between the FDA and EMA labels is the approval for RMS, which includes the spectrum all the way from CIS to SPMS with superimposed relapses. This RMS label was more often granted by the FDA than by the EMA. As apparent from Table 1, 18 compounds have received the RMS label by the FDA, and six under the current EMA label, namely IFNb-1a, ocrelizumab, ofatumumab, cladribine, ponesimod and mitoxantrone (IFNb-1b approved for CIS, RRMS and active SPMS). Interestingly, only the trials for natalizumab, ofatumumab, ocrelizumab, ponesimod, ozanimod and teriflunomide had the definition of RMS listed in their inclusion criteria.
6 Concluding Remarks
As criteria for SPMS are not standardized and have only recently been proposed on scientific grounds [18], it is quite conceivable that the transition phase of MS is even less well defined [46]. Most likely these are patients with reduced but ongoing relapse, and focal inflammatory activity and insidious disability accumulation. Since the latter is not always readily detectable in the standard neurological examination, it is advisable to rely on quantifiable functional scores, such as EDSS, MSFC and its composites 9-HPT, T25FW and SDMT. However, it appears that there is a window of uncertainty that on average lasts for 3 years until a patient is diagnosed with SPMS. Better and standarized criteria may thus help to shorten this latent peroid, for example, the proposed Lorscheider-criteria [18] or tools such as MSProDiscuss. It remains to be shown whether our current understanding of a tipping point event between reduced neuroinflammation and increasing neurodegeneration defining the transition phase is accurate. The concept of PIRA is tempting with regards to measurable disability progression even early in the disease course. However, it does not take into account the potential effects of focal inflammatory activity, for example, new or enlarging lesions on MRI imaging, on disability accumulation.
Scientific evidence for treatment options for the transition phase of MS are sparse; in particular, no trial has yet been conducted to specifically address treatment efficacy in transitioning MS. And even in SPMS only few compounds have been successfully tested in trials specifically designed for SPMS, namely IFNb, mitoxantrone and, most recently, siponimod [45, 48, 52, 79]. However, given the low potency of IFNb and the increased risk for cumulative dose-dependent severe adverse effects for mitoxantrone, these compounds may not be the first options for treating the transition phase of MS. In phase III RCTs for RRMS the proportion of SPMS patients enrolled is small, if not neglectable. Nonetheless, for the FDA the majority of compounds are approved for RMS. Notably, RMS covers “relapsing-remitting disease (RRMS) and active secondary progressive disease”, hence includes the transition period. As per the EMA’s perspective, at the time of the submission of the dossier, the applicant has to apply for a given indication (i.e., RMS, RRMS or SPMS), the acceptability of which is evaluated based on the provided quality, efficacy and safety data package provided by the applicant. For example, siponimod was tested in RRMS patients in its phase II trial BOLD – albeit with MRI surrogate markers as the primary endpoint – and in SPMS patients in its phase III RCT EXPAND [51, 79]. At the time of the submission of the marketing authorisation application, the pharmaceutical company applied for the following indication: treatment of adult patients with secondary progressive multiple sclerosis [80]. Following the evaluation, the indication was restricted for the treatment of adult patients with secondary progressive multiple sclerosis (SPMS) with active disease evidenced by relapses or imaging features of inflammatory activity [81], as explained above. Approval was granted by the FDA for the common RMS label (Table 1). Hence, under the FDA label, patients in the transition phase may be readily treated with siponimod; in contrast, however, according to the EMA label, patients diagnosed with RRMS are not included in the indication for siponimod. Thus, the treating neurologists have to recognize the insididous transition phase in their patients and may have to actively change the diagnosis from RRMS to active SPMS, which may pose an obstactle to some physicians.
While scientific evidence remains sparse for many other DMTs, the majority of these may be used to treat MS patients in the transition phase, at least under the RMS label issued by the FDA (Table 1). In the absence of reliable Class I evidence, those DMTs should be preferred that have shown an effect of disability progression in the respective RCTs and for which post hoc data point towards efficacy in older (> 40 years) and more disabled patients (increased baseline EDSS). Table 2 semi-systematically compiled these data in order to visualize the different strata of scientific rigor available for the approved DMTs. Ocrelizumab, cladribine, fingolimod, ponesimod and ofatumumab may satisfy these criteria; while alemtuzumab does not provide a formal subgroup analysis, it has demonstrated impressive rates of confirmed disability improvement as well as low conversion rates to SPMS during follow-up [76, 82]. Data are equivocal for dimethyl fumarate, teriflunomide, glatiramer acetate and natalizumab, and not sufficient for the remaining IFNb products, diroximel fumarate, and ozanimod.
Interestingly, a recent study, however, did not show evidence that any of the treatment options were able to address disability progression in early SPMS patients [83]. This may be due to the fact that mainly adaptative-mediated focal inflammation is addressed by these treatments, while disease progression in SPMS may be driven by compartimentalized (leptomeningeal or resident) inflammation and non-inflammatory neurodegeneration. Along these lines, a recent meta-analysis demonstrated the role of age as effect modifier for the treatment response and that the efficacy of immunomodulatory DMTs on MS disability strongly decreased with advancing age; in fact this study suggested that beyond the age of 53, DMTs may no longer be efficacious with respect to disability progression [84]. Moreover, DMTs with higher efficacy outperform those with lower efficacy in inhibiting MS disability only for patients younger than 40.5 years in the same study. In this respect, the mean age of MS patients at the transition to SPMS was 44.8 ± 2.2 years [21].
In analogy to the current treatment guidelines for early RRMS it may, therefore, be advisable to readily and actively escalate to a higher DMT in patients deemed to enter the transition phase of MS even in the absence of relapses.
In summary, the insidious transition phase of RRMS to SPMS is not well defined, and RCTs have not been conducted for this condition. As relapse activity in RRMS and SPMS probably share the underlying pathophysiology, the relapse rate reductions in RRMS can be extrapolated on SPMS; extrapolation of the effects of DMT on chronic disability accumulation in RRMS on SPMS is not appropiate because the underlying pathophysiology is likely to differ in these clinical phenotypes. Unequivocal and scientifically sound recommendations are thus not possible. The best data are available for IFNb-1a s.c, IFNb-1b s.c., mitoxantrone and siponimod, the latter being the most modern compound with probably the best risk-to-benefit ratio. All further recommendations can only be derived indirectly from available data from RRMS RCTs. Notably, there are label discrepancies for some DMTs between FDA and EMA (particularly related to the use in SPMS+R as part of the RMS phenotype) that have to be taken into consideration when opting for a specific DMT.
References
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Miller DH, Weinshenker BG, Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14(9):1157–74.
Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647–56.
Giovannoni G, Popescu V, Wuerfel J, et al. Smouldering multiple sclerosis: the “real MS.” Ther Adv Neurol Disord. 2022;15:17562864211066752.
Kappos L, Butzkueven H, Wiendl H, et al. Greater sensitivity to multiple sclerosis disability worsening and progression events using a roving versus a fixed reference value in a prospective cohort study. Mult Scler. 2018;24(7):963–73.
Lorscheider J, Benkert P, Schädelin S, et al., Disability progression unrelated to relapses in relapsing-remitting multiple sclerosis: insights from the Swiss multiple sclerosis cohort study. ECTRIMS Online Library. 2019. 279536(273).
von Wyl V, Benkert P, Lorscheider J et al. Progression independent of relapse activity (PIRA) in relapsing-remitting multiple sclerosis patients on first-line immunomodulatory treatment with fingolimod vs. platform injectables. In: ECTRIMS, 2019. Stockholm.
Graf J, Leussink VI, Soncin G, et al. Relapse-independent multiple sclerosis progression under natalizumab. Brain Commun. 2021;3(4):fcab229.
Lublin FD, Häring DA, Ganjgahi H, et al. How patients with multiple sclerosis acquire disability. Brain. 2022.
Portaccio E, Bellinvia A, Fonderico M, et al. Progression is independent of relapse activity in early multiple sclerosis: a real-life cohort study. Brain. 2022;145(8):2796–805.
Kappos L, Wolinsky JS, Giovannoni G, et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020;77(9):1132–40.
European Medicines Agency Science Medicines Health. (2015, March 26). Guideline on clinical investigation of medicinal products for the treatment of Multiple Sclerosis. European Medicines Agency Science Medicines Health. Retrieved September 18, 2022, from https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-multiple-sclerosis_en-0.pdf.
Barzegar M, Najdaghi S, Afshari-Safavi A, et al. Early predictors of conversion to secondary progressive multiple sclerosis. Mult Scler Relat Disord. 2021;54: 103115.
Tremlett H, Yinshan Z, Devonshire V. Natural history of secondary-progressive multiple sclerosis. Mult Scler. 2008;14(3):314–24.
Abdelhak A, Weber MS, Tumani H. Primary progressive multiple sclerosis: putting together the puzzle. Front Neurol. 2017;8:234.
Rice CM, Cottrell D, Wilkins A, et al. Primary progressive multiple sclerosis: progress and challenges. J Neurol Neurosurg Psychiatry. 2013;84(10):1100–6.
Butzkueven H, Chapman J, Cristiano E, et al. MSBase: an international, online registry and platform for collaborative outcomes research in multiple sclerosis. Mult Scler. 2006;12(6):769–74.
Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain. 2016;139(Pt 9):2395–405.
Kopp TI, Bramow S, Illes Z, et al. Application of definitions for conversion to secondary progressive MS in a Danish nationwide population. Mult Scler Relat Disord. 2021;56: 103319.
Boyko A, Therapontos C, Horakova D, et al. Approaches and challenges in the diagnosis and management of secondary progressive multiple sclerosis: a Central Eastern European perspective from healthcare professionals. Mult Scler Relat Disord. 2021;50: 102778.
Katz Sand I, Krieger S, Farrell C, et al. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler. 2014;20(12):1654–7.
Rojas JI, Patrucco L, Alonso R, et al. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis: multicenter study in Argentina. Mult Scler. 2021;27(4):579–84.
Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(Pt 5):1175–89.
Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. 2018;9:3116.
Williams T, Zetterberg H, Chataway J. Neurofilaments in progressive multiple sclerosis: a systematic review. J Neurol. 2021;268(9):3212–22.
Invernizzi P, Bertolasi L, Bianchi MR, et al. Prognostic value of multimodal evoked potentials in multiple sclerosis: the EP score. J Neurol. 2011;258(11):1933–9.
Leocani L, Rovaris M, Boneschi FM, et al. Multimodal evoked potentials to assess the evolution of multiple sclerosis: a longitudinal study. J Neurol Neurosurg Psychiatry. 2006;77(9):1030–5.
Margaritella N, Mendozzi L, Garegnani M, et al. Sensory evoked potentials to predict short-term progression of disability in multiple sclerosis. Neurol Sci. 2012;33(4):887–92.
Schlaeger R, D’Souza M, Schindler C, et al. Prediction of MS disability by multimodal evoked potentials: investigation during relapse or in the relapse-free interval? Clin Neurophysiol. 2014;125(9):1889–92.
Abalo-Lojo JM, Limeres CC, Gómez MA, et al. Retinal nerve fiber layer thickness, brain atrophy, and disability in multiple sclerosis patients. J Neuroophthalmol. 2014;34(1):23–8.
Martinez-Lapiscina EH, Arnow S, Wilson JA, et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: a cohort study. Lancet Neurol. 2016;15(6):574–84.
Ratchford JN, Saidha S, Sotirchos ES, et al. Active MS is associated with accelerated retinal ganglion cell/inner plexiform layer thinning. Neurology. 2013;80(1):47–54.
Valsasina P, Rocca MA, Horsfield MA, et al. Regional cervical cord atrophy and disability in multiple sclerosis: a voxel-based analysis. Radiology. 2013;266(3):853–61.
Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol. 2018;83(2):210–22.
Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653–66.
Gil-Perotin S, Alcalá C, Pérez-Miralles FC, et al. Silent progression or bout onset progressive multiple sclerosis? Ann Neurol. 2019;86(3):472.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–52.
Amato MP, Prestipino E, Bellinvia A. Identifying risk factors for cognitive issues in multiple sclerosis. Expert Rev Neurother. 2019;19(4):333–47.
Zipoli V, Goretti B, Hakiki B, et al. Cognitive impairment predicts conversion to multiple sclerosis in clinically isolated syndromes. Mult Scler. 2010;16(1):62–7.
European Medicines Agency Science Medicines Health. (2020, February 26). Qualification opinion on Multiple Sclerosis Clinical Outcome Assessment (MSCOA). European Medicines Agency Science Medicines Health. Retrieved September 18, 2022, from https://www.ema.europa.eu/en/documents/other/qualification-opinion-multiple-sclerosisclinical-outcome-assessment-mscoa_en.pdf.
Polman CH, Rudick RA. The multiple sclerosis functional composite: a clinically meaningful measure of disability. Neurology. 2010;74(Suppl 3):S8-15.
Wolinsky JS, Arnold DL, Brochet B, et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):998–1009.
Ziemssen T, Giovannoni G, Alvarez E, et al. Multiple sclerosis progression discussion tool usability and usefulness in clinical practice: cross-sectional, web-based survey. J Med Internet Res. 2021;23(10): e29558.
Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352(9139):1498–504.
Randomized controlled trial of interferon- beta-1a in secondary progressive MS: clinical results. Neurology. 2001;56(11):1496–504.
Freedman MS, Brod S, Singer BA, et al. Clinical and MRI efficacy of sc IFN β-1a tiw in patients with relapsing MS appearing to transition to secondary progressive MS: post hoc analyses of PRISMS and SPECTRIMS. J Neurol. 2020;267(1):64–75.
Plosker GL. Interferon-β-1b: a review of its use in multiple sclerosis. CNS Drugs. 2011;25(1):67–88.
Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. 1998;352(9139):1491–7.
Panitch H, Miller A, Paty D, et al. Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology. 2004;63(10):1788–95.
Kappos L, Li DK, Stüve O, et al. Safety and efficacy of siponimod (BAF312) in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the phase 2 BOLD study. JAMA Neurol. 2016;73(9):1089–98.
Selmaj K, Li DK, Hartung HP, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756–67.
Hartung HP, Gonsette R, König N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018–25.
Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221–34.
Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–20.
Wolinsky JS, Engmann NJ, Pei J, et al. An exploratory analysis of the efficacy of ocrelizumab in patients with multiple sclerosis with increased disability. Mult Scler J Exp Transl Clin. 2020;6(1):2055217320911939.
European Medicines Agency Science Medicines Health. (2017, November 9). Assessment report Ocrevus International non-proprietary name: ocrelizumab Procedure No. EMEA/H/C/004043/0000. European Medicines Agency Science Medicines Health. Retrieved September 18, 2022, from https://www.ema.europa.eu/en/documents/assessment-report/ocrevus-epar-public-assessment-report_en.pdf.
Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–57.
Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab vs teriflunomide in relapsing multiple sclerosis: analysis of no evidence of disease activity (NEDA-3) from ASCLEPIOS I and II trials. Eur J Neurol. 2020;27:1268–307.
Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416–26.
Rammohan K, Giovannoni G, Comi G, et al. Cladribine tablets for relapsing-remitting multiple sclerosis: efficacy across patient subgroups from the phase III CLARITY study. Mult Scler Relat Disord. 2012;1(1):49–54.
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–97.
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107.
Viglietta V, Miller D, Bar-Or A, et al. Efficacy of delayed-release dimethyl fumarate in relapsing-remitting multiple sclerosis: integrated analysis of the phase 3 trials. Ann Clin Transl Neurol. 2015;2(2):103–18.
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–303.
Miller AE, O’Connor P, Wolinsky JS, et al. Pre-specified subgroup analyses of a placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis. Mult Scler. 2012;18(11):1625–32.
Derfuss T, Ontaneda D, Nicholas J, et al. Relapse rates in patients with multiple sclerosis treated with fingolimod: Subgroup analyses of pooled data from three phase 3 trials. Mult Scler Relat Disord. 2016;8:124–30.
Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–84.
DiBernardo A, Turkoz I. Ponesimod demonstrated increased clinical benefit over teriflunomide in early disease subgroup compared with overall population. E-poster at the Congress of the European Committee for Treatment Research in Multiple Sclerosis (ECTRIMS) 13–15 October 2021.
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
Kapoor R, Ho PR, Campbell N, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018;17(5):405–15.
Hutchinson M, Kappos L, Calabresi PA, et al. The efficacy of natalizumab in patients with relapsing multiple sclerosis: subgroup analyses of AFFIRM and SENTINEL. J Neurol. 2009;256(3):405–15.
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–28.
Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–39.
Coles AJ, Fox E, Vladic A, et al. Alemtuzumab versus interferon β-1a in early relapsing-remitting multiple sclerosis: post-hoc and subset analyses of clinical efficacy outcomes. Lancet Neurol. 2011;10(4):338–48.
Coles AJ, Arnold DL, Bass AD, et al. Efficacy and safety of alemtuzumab over 6 years: final results of the 4-year CARE-MS extension trial. Ther Adv Neurol Disord. 2021;14:1756286420982134.
Horáková D, Boster A, Bertolotto A, et al. Proportion of alemtuzumab-treated patients converting from relapsing-remitting multiple sclerosis to secondary progressive multiple sclerosis over 6 years. Mult Scler J Exp Transl Clin. 2020;6(4):2055217320972137.
Johnson KP, Brooks BR, Cohen JA, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1998;50(3):701–8.
Liu C, Blumhardt LD. Benefits of glatiramer acetate on disability in relapsing-remitting multiple sclerosis. An analysis by area under disability/time curves. The Copolymer 1 Multiple Sclerosis Study Group. J Neurol Sci. 2000;181(1–2):33–7.
Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–73.
European Medicines Agency Science Medicines Health. (2019, November 14). Assessment report Mayzent International non-proprietary name: siponimod Procedure No. EMEA/H/C/004712/0000. European Medicines Agency Science Medicines Health. Retrieved September 18, 2022, from https://www.ema.europa.eu/en/documents/assessment-report/mayzent-epar-public-assessment-report_en.pdf.
European Medicines Agency Science Medicines Health. Annex I-III, labelling and package leaflet. European Medicines Agency Science Medicines Health. Retrieved September 18, 2022, from https://www.ema.europa.eu/en/documents/product-information/mayzent-epar-product-information_en.pdf.
Riera R, Porfírio GJ, Torloni MR. Alemtuzumab for multiple sclerosis. Cochrane Database Syst Rev. 2016;4(4):Cd011203.
Roos I, Leray E, Casey R, et al. Effects of high- and low-efficacy therapy in secondary progressive multiple sclerosis. Neurology. 2021;97(9):e869–80.
Weideman AM, Tapia-Maltos MA, Johnson K, et al. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. 1996;39(3):285–94.
Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993. 43(4):655–61.
Kappos L, Freedman MS, Polman CH, et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5-year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol. 2009;8(11):987–97.
Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014;13(7):657–65.
Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9700):1503–11.
Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–56.
Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2014;20(6):705–16.
The U.S. Food and Drug Administration Highlights of Prescribing Information.
Berger AA, Sottosanti ER, Winnick A, et al. Monomethyl fumarate (MMF, Bafiertam) for the treatment of relapsing forms of multiple sclerosis (MS). Neurol Int. 2021;13(2):207–23.
Naismith RT, Wundes A, Ziemssen T, et al. Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. CNS Drugs. 2020;34(2):185–96.
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–56.
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15.
Kappos L, Fox RJ, Burcklen M, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: a randomized clinical trial. JAMA Neurol. 2021;78(5):558–67.
Cohen JA, Comi G, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019;18(11):1021–33.
Comi G, Kappos L, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019;18(11):1009–20.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. There was no funding involved in the preparation of the manuscript.
Conflict of interest
Nikolaos G. Dimitriou: no conflicts of interest to disclose. Sven G. Meuth: receives honoraria for lecturing and travel expenses for attending meetings from Almirall, Amicus Therapeutics Germany, Bayer Health Care, Biogen, Celgene, Diamed, Genzyme, MedDay Pharmaceuticals, Merck Healthcare, Novartis, Novo Nordisk, ONO Pharma, Roche, Sanofi-Aventis, Chugai Pharma, QuintilesIMS und Teva. His research is funded by the German Ministry for Education and Research (BMBF), Deutschen Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, German Academic Exchange Service, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, German Foundation Neurology and by Almirall, Amicus Therapeutics Germany, Biogen Idec, Diamed, Fresenius Medical Care, Genzyme, Merck Healthcare, Novartis, ONO Pharma, Roche, und Teva. Elena H Martinez-Lapiscina: no conflicts of interest to disclose. The views expressed in this article are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties. Til Menge: personal fees from Biogen, BMS, Novartis, Teva, Roche, Merck Serono; non-financial support from Biogen, Merck Serono, Roche. Philipp Albrecht: received grants and non-financial support from Biogen; grants, personal fees and non-financial support from Allergan / Abbvie; personal fees and non-financial support from Bayer; personal fees and non-financial support from Sanofi Genzyme; grants, personal fees and non-financial support from Merck; grants, personal fees and non-financial support from Merz Pharmaceuticals; grants, personal fees and non-financial support from Novartis; grants, personal fees and non-financial support from Roche; grants, personal fees and non-financial support from Teva; and grants, personal fees and non-financial support from Ipsen, outside the submitted work.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
NGD: first draft of manuscript and writing of manuscript. SGM: review and revision of the text. EHM-L: review and revision of the text. TM: first draft of manuscript and writing of manuscript. PA: First draft of manuscript and writing of manuscript. All authors have read and approve the final version of the manuscript, and agree to be accountable for the work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Dimitriou, N.G., Meuth, S.G., Martinez-Lapiscina, E.H. et al. Treatment of Patients with Multiple Sclerosis Transitioning Between Relapsing and Progressive Disease. CNS Drugs 37, 69–92 (2023). https://doi.org/10.1007/s40263-022-00977-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-022-00977-3