
Abstract
Background and Objective
Lorlatinib is a third-generation tyrosine kinase inhibitor approved for the second-line treatment of patients with advanced anaplastic lymphoma kinase-positive non-small cell lung cancer. Lorlatinib is metabolized by cytochrome P450 (CYP) 3A and contraindicated with strong CYP3A inducers because of significant transaminase elevation. This phase I, open-label, two-period study evaluated the impact of a moderate CYP3A inducer, modafinil, on the safety and pharmacokinetics of lorlatinib.
Methods
Healthy participants received single-dose oral lorlatinib (50 mg [n = 2], 75 mg [n = 2], or 100 mg [n = 2 + 10 in an expanded cohort]) in Period 1 followed by modafinil 400 mg/day (days 1–19) and single-dose lorlatinib (day 15, same dose as previous) both orally in Period 2. Blood samples were collected for 120 h after each dose of lorlatinib.
Results
Of 16 participants, ten completed the study; six participants, all in the expanded 100-mg cohort, discontinued because of adverse events during the modafinil lead-in dosing period. Single doses of lorlatinib 50–100 mg were well tolerated when administered alone and in the presence of steady-state modafinil. Of the ten participants who completed the study, all had transaminase values within normal limits during the combination of lorlatinib with modafinil. The ratios of the adjusted geometric means (90% confidence interval) for lorlatinib area under the plasma concentration–time profile extrapolated to infinity and maximum plasma concentration were 76.69% (70.15–83.83%) and 77.78% (65.92–91.77), respectively, when lorlatinib 100 mg was co-administered with steady-state modafinil compared with lorlatinib administration alone.
Conclusion
Lorlatinib 100 mg may be safely co-administered with moderate CYP3A inducers.
Clinical Trial Registration
ClinicalTrials.gov NCT03961997; registered 23 May, 2019.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Single doses of lorlatinib 50–100 mg were well tolerated when administered alone or with steady-state modafinil, a moderate cytochrome P450 3A inducer, with no clinically significant signal of hepatotoxicity. |
Co-administration of lorlatinib 100 mg in the presence of steady-state modafinil decreased the area under the plasma concentration–time profile extrapolated to infinity and maximum plasma concentration by approximately 23% and 22%, respectively, compared with lorlatinib given alone. |
Lorlatinib 100 mg may be safely co-administered with moderate CYP3A inducers. |
1 Introduction
Lorlatinib (PF-06463922) is a potent and selective third-generation inhibitor of the anaplastic lymphoma kinase (ALK) and c-ROS oncogene 1 kinases, that also inhibits ALK resistance mutations responsible for resistance to crizotinib [1,2,3]. In the ongoing, phase I/II, clinical study B7461001 (NCT01970865), lorlatinib demonstrated substantial overall and intracranial activity with a favorable safety and tolerability profile, in patients with ALK-positive non-small cell lung cancer [4, 5]. Lorlatinib is currently approved at a 100-mg once-daily (QD) dose in several countries, including the USA, Japan, and the European Union, for the second-line treatment of patients with advanced ALK-positive non-small cell lung cancer.
The metabolism of lorlatinib is primarily by cytochrome P450 (CYP) 3A4 and uridine diphosphate-glucuronosyltransferase (UGT) 1A4, with minor contributions from CYP2C8, CYP2C19, CYP3A5, and UGT1A3. The most abundant circulating metabolite in human plasma, PF-06895751 (M8), is pharmacologically inactive and likely involves multiple biotransformation steps, including CYP3A4/5. In the previous drug–drug interaction study B7461011 (NCT02804399) [6], all 12 participants had 3-fold to 25-fold increases in aspartate aminotransferase (AST) or alanine aminotransferase (ALT) after receiving a single dose of lorlatinib (100 mg) with rifampin (600 mg QD) after multiple doses of rifampin, with five being hospitalized and reported as serious adverse events (AEs). Transaminase levels all returned to normal levels within a median of 15 days after discontinuation of rifampin dosing. Based on the findings from that study, lorlatinib dosing in combination with strong CYP3A inducers is contraindicated on the drug label.
This study was conducted to evaluate whether similar AST and ALT elevations may also occur in those receiving lorlatinib with a moderate CYP3A inducer. Modafinil 400 mg QD has been previously used as a perpetrator drug for eliciting moderate CYP3A induction in pharmacokinetic (PK) drug interaction studies [7,8,9]. As lorlatinib is a substrate for CYP3A, concomitant administration of multiple doses of the moderate CYP3A inducer modafinil along with lorlatinib was expected to decrease systemic plasma exposure to lorlatinib. The primary objective of this study was to assess the safety and tolerability of a single dose of lorlatinib 100 mg in healthy adult participants when administered alone and following multiple doses of modafinil. The secondary objective of this study was to assess the effect of the moderate CYP3A inducer modafinil on lorlatinib pharmacokinetics. This study was designed to ensure participant safety, with sequential dose escalation of lorlatinib (starting with a 50-mg single dose through the approved starting 100-mg lorlatinib dose), and with sentinel dosing such that only one participant at each lorlatinib dose was initially followed for safety before enrolling more participants in that dosing cohort.
2 Methods
2.1 Study Design and Participants
This was a phase I, open-label, two-period, two-treatment, fixed-sequence study in healthy participants (B7461026; NCT03961997) conducted between 22 August, 2019 and 9 December, 2019. The schedule of dosing and assessments for Period 1 is presented in Table S1 of the Electronic Supplementary Material (ESM). Each participant received a single oral dose of lorlatinib 50, 75, or 100 mg alone followed by a washout period of at least 19 days prior to the second dose of lorlatinib. The first lorlatinib dose was given in the morning at approximately 08:00 (± 2) hours on day 1 following an overnight 10-hour fast. The schedule of dosing and assessments for Period 2 is presented in Table S2 of the ESM. After the previous washout, participants received oral doses of modafinil 400 mg (4 × 100-mg tablets) QD on days 1–19. Modafinil doses on days 1, 8, and 14–19 were administered at approximately 08:00 (± 2) hours, in the presence of Pfizer Clinical Research Unit staff. Doses of modafinil on days 2–7 and days 9–13 were self-administered at home, with dosing details recorded in a diary provided to the participant. On days 1–14 and days 16–19, administration of modafinil was at least 1 hour before or at least 2 h after a meal. On day 15, modafinil and lorlatinib were administered within 5 minutes of each other after an overnight fast of at least 10 hours at approximately 08:00 (±2) hours.
Cohorts 1–3 included two participants each and Cohort 4 included ten participants. Cohorts 1–3 were enrolled sequentially with the lorlatinib 50-mg single-dose cohort first (Cohort 1), followed by the 75-mg cohort (Cohort 2), and then finally by the 100-mg cohort (Cohort 3). In Cohorts 1–3, the second participants received their lorlatinib Period 2 dose in combination with modafinil only after the first participants were 72 h past their lorlatinib Period 2 dose with no safety concerns raised. All Cohort 4 participants (lorlatinib 100-mg single-dose expanded cohort) received their lorlatinib Period 2 dose after the last participant in Cohort 3 was 72 h past their lorlatinib Period 2 dose with no safety concerns.
Healthy female participants of non-childbearing potential and/or male participants aged between 18 and 55 years, and with adequate renal and liver function, were to be enrolled. Adequate renal function included an estimated creatinine clearance ≥ 90 mL/min and adequate liver function included AST, ALT, alkaline phosphatase, and total bilirubin all within normal limits. Key exclusion criteria included evidence or a history of clinically significant hematological, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, neurological, or allergic disease; current use of prescription or non-prescription drugs and/or dietary or herbal supplements (excluding paracetamol ≤ 1 g/day) within 7 days (or five half-lives, whichever was longer); previous administration of lorlatinib within 30 days (or five half-lives, whichever was longer); previous treatment with an investigational drug within 30 days (or five half-lives, whichever was longer); previous treatment with lorlatinib; use of strong CYP3A inhibitors or inducers within 14 days prior to the first dose of study treatment. A comprehensive list of the selection criteria is provided in Table S3 of the ESM.
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonisation Good Clinical Practice Guidelines. In addition, all local regulatory requirements were followed. All participants provided written informed consent before undergoing any study procedures.
2.2 Safety Assessments
Adverse events, laboratory parameters, vital signs, and electrocardiograms were evaluated at screening and across both periods in all cohorts. Urinalysis and blood samples (10 mL) for safety laboratory assessments were collected following at least a 4-h fast at screening and on day − 1, days 2–6 of Period 1, and days 14, 15 (prior to lorlatinib dosing and 12 h post-dosing), and days 16–20 (prior to modafinil dosing on days 16–19 and 12 hours post-dosing on days 16 and 17) of Period 2.
2.3 PK Assessments
Blood samples (6 mL) to provide approximately 2.4 mL of plasma for determination of plasma concentrations of lorlatinib and its metabolite (M8) were collected into tubes containing dipotassium ethylenediaminetetraacetic acid. Blood samples were collected from participants pre-dose and 0.5, 1, 1.5, 2, 4, 6, 12, 24, 36, 48, 60, 72, 96, and 120 hours post-oral dosing of lorlatinib. Samples collected for the measurement of plasma concentrations of lorlatinib and M8 were analyzed by Covance Bioanalytical Services (Shanghai, China) using a validated liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) method with a lower limit of quantification (LLOQ) of 2.50 ng/mL and 1.00 ng/mL, respectively (more details are provided in Sect. S2.3 of the ESM).
Plasma PK parameters for lorlatinib and its metabolite (M8) were estimated for each participant and treatment period using a non-compartmental analysis conducted in Pfizer internal software, eNCA. Samples below the LLOQ were set to 0 ng/mL for the PK analysis. Maximum plasma concentration (Cmax) and time of peak concentration were determined from observed data. The area under the curve (AUC) of the plasma concentration–time profile from time 0 to the time of last measured plasma concentration (AUClast) was estimated using the linear/log trapezoidal method. The AUC from time 0 extrapolated to infinite time (AUCinf) was calculated as AUClast + (Clast/kel), where Clast was the predicted plasma concentration at the last quantifiable time point estimated from the log-linear regression analysis and kel was the terminal phase rate constant calculated by linear regression of the log-linear concentration–time curve. Terminal plasma half-life was calculated as Loge(2)/kel. Apparent oral clearance was calculated as dose/AUCinf and apparent volume of distribution was calculated as dose/(AUCinf × kel).
2.4 Other Assessments
Plasma samples for cholesterol and 4β-hydroxycholesterol and urine samples for cortisol and 6β-hydroxycortisol were collected pre-dose on days 1, 8, and 15 of Period 2. Plasma samples were analyzed by Q2 Solutions (Ithaca, NY, USA) for cholesterol and 4β-hydroxycholesterol using a validated LC-MS/MS method with a LLOQ of 500 μg/mL and 3.00 ng/mL, respectively. Urine samples were analyzed by PPD (Richmond, VA, USA) for cortisol and 6β-hydroxycortisol using a validated high-performance LC-MS/MS method, both with a LLOQ of 1.00 ng/mL. Plasma 4β-hydroxycholesterol/cholesterol molar ratios and urine 6β-hydroxycortisol/cortisol concentration ratios were calculated as estimates of CYP3A induction. Additional details on bioanalytical assays are provided in Sect. S2.4 of the ESM.
2.5 Statistical Analysis
Three analysis populations were prespecified: (1) the PK concentration population included all participants who received the study drug and had at least one lorlatinib concentration in at least one treatment period; (2) the PK parameter analysis population included all participants who received the study drug and had at least one of the lorlatinib PK parameters of interest in one or more treatment periods; and (3) the safety analysis population included all participants who received at least one dose of either study drug.
This study was not prospectively powered to detect a specified change in lorlatinib exposure (AUCinf or Cmax) because its primary objective was to evaluate safety. Two participants each for 50 mg and 75 mg and 12 participants for 100 mg were intended to be recruited. It was determined that a sample size of 12 participants at the highest lorlatinib 100-mg dose (combined Cohorts 3 and 4) would provide a 90% confidence interval (CI) for the difference between treatments of ± 0.053 and ± 0.090 on the natural log scale for AUCinf and Cmax, respectively, with 90% coverage probability. This calculation was based on estimates of within-participant standard deviations of 0.082 and 0.138 for AUCinf and Cmax on the natural logarithm scale, respectively, established from prior clinical studies of lorlatinib in healthy participants.
Natural log-transformed AUCinf, AUClast, and Cmax for lorlatinib were analyzed using a mixed-effects model, with treatment as the fixed effect and the participant as a random effect, and estimates of the adjusted mean differences (test–reference) and corresponding 90% CIs were obtained. Lorlatinib alone was the reference treatment and lorlatinib with modafinil was the test treatment. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (test/reference) and 90% CIs for the ratios.
The lorlatinib and M8 PK parameters AUCinf, Cmax, AUClast, time of peak concentration, and half-life were summarized descriptively by treatment. For lorlatinib AUCinf and Cmax, individual participant parameters were plotted by treatment. Median profiles of the concentration–time data were plotted by treatment; the nominal PK sampling time was used for summary statistics and median plots by sampling time. Adverse events were tabulated descriptively by dose and treatment.
3 Results
3.1 Participants
A total of 16 healthy participants, all white and male, with a median age of 31.5 years and a median weight and body mass index of 72.9 kg and 23.1 kg/m2, respectively, were enrolled and treated (Table 1). Ten participants completed the study, including all six participants (two each) in Cohorts 1–3 (lorlatinib 50 mg, 75 mg, and 100 mg, respectively) and four in Cohort 4 (lorlatinib 100 mg). Six participants in Cohort 4 discontinued from the study because of AEs during the administration of modafinil 400 mg QD alone. All 16 participants received one or more doses of the study drug and, with evaluable PK data for lorlatinib and its metabolite M8, were included in the safety and PK analysis populations as well as in the PK concentration population.
3.2 Safety
In Period 1, all 16 participants received a single dose of lorlatinib alone. In Period 2, all 16 participants initially received modafinil 400 mg QD alone: ten completed the study with 14 days of modafinil QD dosing on days 1–14, concomitant single-dose lorlatinib with modafinil 400 mg on day 15, and then four additional doses of modafinil 400 mg QD on days 16–19 (two participants in Cohort 1, two in Cohort 2, and six in Cohorts 3 and 4 combined). Six participants, all in Cohort 4, received 4–7 days of QD dosing of modafinil prior to their discontinuation from the study.
A total of 121 treatment-emergent AEs were reported during the study, 114 of which were considered treatment related by the investigator (Table S4 of the ESM). All AEs were mild to moderate and resolved by the end of the study. There were no serious or severe AEs reported. Most AEs (96 out of 121) were reported following modafinil 400 mg QD alone. The most frequently reported AEs were palpitations, decreased appetite, insomnia, headache, anxiety, nausea, and fatigue, all of which were considered treatment related. Of 44 moderate AEs, 43 were observed while participants were taking modafinil alone, and 42 were considered treatment related. The most frequently reported moderate treatment-related AEs were palpitations, decreased appetite, insomnia, and anxiety.
In Cohorts 3 and 4 combined (lorlatinib 100 mg), 12 participants received single-dose lorlatinib 100 mg alone in Period 1. A total of 15 AEs, all mild in severity, were reported by six participants during Period 1. The most frequently reported system organ class was gastrointestinal disorders reported by three participants and the only preferred term reported by more than one participant was abdominal pain (by two participants). Of the six participants in Cohorts 3 and 4 who received co-administration of single-dose lorlatinib 100 mg with modafinil 400 mg, four reported a total of seven treatment-related AEs, all mild in severity. The most frequently reported system organ class was psychiatric disorders reported by three participants and the only preferred term reported by more than one participant was euphoric mood (in two participants). The other AEs were insomnia, feeling of relaxation, diarrhea, nausea, decreased appetite, and photophobia, each in one participant (all considered treatment related, except for photophobia).
As noted earlier, six participants, all in Cohort 4, permanently discontinued from the study in Period 2 because of AEs when receiving modafinil 400 mg QD alone (prior to receiving combination dosing with lorlatinib). All 37 AEs reported by these participants were moderate in severity at discontinuation, resolved following modafinil discontinuation, and were considered modafinil related. One participant in Cohort 1 receiving modafinil 400 mg QD alone had a temporary drug interruption (skipped one modafinil dose on day 11 of Period 2) because of mild diarrhea and moderate vomiting, both of which were considered treatment related.
All 12 participants in Cohorts 3 and 4 combined (lorlatinib 100 mg) had liver function test values within normal limits at baseline and during single-dose lorlatinib 100-mg treatment, with one exception (Fig. 1). One participant had AST and ALT values that slightly exceeded the upper limit of normal (ULN; peak values < 1.4 × ULN) on study days 5 and 6 (Period 1 day 5 and Period 1 day 6/Period 2 day 1), but returned to normal on study day 19 (Period 2 day 14; prior to start of combination lorlatinib and modafinil dosing) and remained normal until study completion (Fig. 1). In the six participants in Cohorts 3 and 4 combined who completed the study, minor increases in AST and ALT values following co-administration of single-dose lorlatinib 100 mg with modafinil 400 mg QD were noted; however, all remained within normal ranges (Fig. 1). Most participants reached peak levels around 36 h post-lorlatinib dose, with median (range) AST values of 30.5 (15.0–39.0) U/L (ULN = 40), and ALT values of 32.5 (15.0–45.0) U/L (ULN = 49), respectively. Alkaline phosphatase and total bilirubin results were all within normal limits.

Line plots of individual participant and median liver function tests: a aspartate aminotransferase (AST), b alanine aminotransferase (ALT), c alkaline phosphatase (ALP), and d total bilirubin, for the 100-mg cohort (Cohorts 3 and 4 combined). The red dashed lines on the figures represent the upper limit of normal (ULN) for AST (40 U/L), ALT (49 U/L), ALP (111 U/L), and total bilirubin (1.2 mg/dL), respectively. Study day 1 = Period 1 day 1; day 6 = Period 1 day 6/Period 2 day 1; day 20 = Period 2 day 15. Age-adjusted ULN of 127 U/L for ALP was applicable to a 55-year-old participant who completed the study in Cohort 4. QD once daily
All four participants in Cohorts 1 and 2 (lorlatinib 50 mg and 75 mg, respectively) had normal liver function test values during the study except for one participant in Cohort 2 whose total bilirubin value slightly exceeded ULN (< 1.2 × ULN) once on study day 19 (Period 2 day 14) during modafinil 400-mg QD treatment. There were no clinically significant findings in other laboratory tests, vital signs, and electrocardiograms.
3.3 Lorlatinib Pharmacokinetics
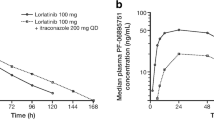
Median lorlatinib plasma concentrations across all cohorts were lower in the presence of steady-state modafinil 400 mg QD than observed when lorlatinib was administered alone (Fig. 2 for the 100-mg cohort, Fig. S1 of the ESM for all cohorts). Lorlatinib AUCinf and Cmax values were likewise lower in the presence of steady-state modafinil than when lorlatinib was administered alone (Table 2 for the 100 mg cohort, Table S5 of the ESM for all cohorts). This corresponded to the apparent oral clearance of lorlatinib at 100 mg being higher (16.84 L/h geometric mean) when co-administered with steady-state modafinil vs when lorlatinib was administered alone (12.85 L/h).

a Plasma lorlatinib and b metabolite (M8) concentration–time profiles for the 100-mg cohort (Cohorts 3 and 4 combined) following administration of a single oral dose of lorlatinib alone and in the presence of steady-state modafinil. QD once daily
The ratios of the adjusted geometric means (90% CI) for lorlatinib AUCinf and Cmax were 76.69% (70.15–83.83) and 77.78% (65.92–91.77), respectively, when lorlatinib was co-administered with steady-state modafinil compared with lorlatinib administration alone (Table 3). Median time of peak concentration for lorlatinib 100 mg was 1.5 hours both in the presence and absence of modafinil. Mean estimates of the plasma half-life for lorlatinib 100 mg were similar (22.5 hours and 23.1 hours) in the absence and presence of modafinil, respectively.
Median M8 plasma concentrations, and AUCinf and Cmax values, in the 100-mg cohort were also lower in the presence of steady-state modafinil than observed when lorlatinib was administered alone (Fig. 2, Table 2). The metabolite-to-parent ratio for AUCinf and the metabolite-to-parent ratio for Cmax were 1.711 and 0.3233, respectively, following administration of lorlatinib 100 mg alone, and 1.237 and 0.2639, respectively, following co-administration of lorlatinib 100 mg and steady-state modafinil. Inter-participant variability for lorlatinib 100 mg and M8 AUCinf and Cmax geometric mean values were similar when lorlatinib was co-administered with steady-state modafinil compared to its administration alone.
3.4 Modafinil CYP3A Induction
Plasma 4β-hydroxycholesterol/cholesterol ratios increased over time in most participants, with median (range) values (molar ratios × 10-5) of 0.99 (0.62–1.72), 1.70 (0.81–4.07), and 2.04 (0.84–3.22) on Period 2 days 1, 8, and 15, respectively, for Cohorts 3 and 4 combined (Fig. 3, Table S6 of the ESM). Urine 6β-hydroxycortisol/cortisol concentration ratios did not consistently change over time, with median (range) values of 9.7 (2.4–31.3), 9.7 (5.2–14.8), and 9.7 (2.7–19.6) on Period 2 days 1, 8, and 15, respectively (Fig. S2 of the ESM).

Matchstick plots for plasma 4β-hydroxycholesterol/cholesterol ratios (molar ratios × 10− 5) pre-modafinil and post-modafinil once-daily treatments. Only participants who had at least one plasma 4β-hydroxycholesterol/cholesterol ratio are represented. Data represent individual participant values (participants 1 and 2 were in Cohort 1 [lorlatinib 50 mg], participants 3 and 4 were in Cohort 2 [lorlatinib 75 mg], and participants 5–16 were in Cohorts 3 and 4 combined (lorlatinib 100 mg])
4 Discussion
Single doses of lorlatinib 50–100 mg were well tolerated when administered alone and co-administered in the presence of steady-state modafinil 400 mg QD, a moderate CYP3A inducer. No clinically significant signal of hepatotoxicity was identified for this co-administration. Of the ten participants who completed the study (two participants each receiving lorlatinib 50 mg and 75 mg, and six receiving lorlatinib 100 mg), all had AST and ALT values within normal limits during combination treatment with modafinil. These results are in sharp contrast to the observations in the prior drug–drug interaction study with the combination of single-dose lorlatinib 100 mg and rifampin 600 mg QD in which all 12 participants had significant asymptomatic AST and/or ALT increases following the co-administration with rifampin, a strong pan-inducer of many enzymes including CYP3A [6]. In that study, grade 4 elevations (as per National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03) in AST or ALT occurred in six participants, grade 3 elevations in four participants, and grade 2 elevations in one participant. The AST/ALT elevations appeared within 3 days following the co-administration of lorlatinib and rifampin, and as a result, rifampin treatment was discontinued and participants did not receive the planned additional three daily doses of rifampin 600 mg QD. Five participants with particularly high AST and ALT values were hospitalized for observation, and these events were noted as serious AEs and considered treatment related by the investigator. While none of the participants in the current study had AST/ALT levels out of the normal range, minor temporal increases in median AST and ALT values (10 and 15 U/L, respectively) were observed in the six participants following co-administration of single-dose lorlatinib 100 mg and modafinil; this indicated that there is a mechanistic change in AST/ALT but substantially attenuated compared with what was seen with rifampin, thus the magnitude of change was not considered clinically meaningful. No serious or severe AEs were reported in the current study. All AEs reported following single-dose lorlatinib and single-dose lorlatinib co-administration with modafinil were mild in severity, except one moderate AE. None of the AEs that occurred following co-administration of steady-state modafinil with single-dose lorlatinib resulted in study discontinuation, or indicated hepatotoxicity or other safety concerns, and there were no other clinically significant findings. Based on these results, the overall assessment from this study is that lorlatinib can be safely administered with moderate CYP3A inducers.
Six participants in the lorlatinib 100-mg cohorts discontinued from the study, but this happened because of modafinil-related AEs during the 14-day modafinil induction phase prior to receiving combination dosing with lorlatinib. It is our speculation that the simultaneous enrollment of ten participants in Cohort 4 may have possibly contributed to the cluster discontinuations in that cohort. Multiple AEs resulted in each of the discontinuations, all moderate in severity and considered related to modafinil by the investigator. The most common AEs were anxiety, insomnia, and decreased appetite. Interactions between participants could have elevated participant anxiety, which may have resulted in increased numbers of AEs and decreased tolerability. However, both lorlatinib treatments (alone and co-administration with modafinil) appeared to be less impacted as no participants discontinued the study because of the AEs following treatments involving lorlatinib.
The PK results of this study indicated that co-administration of a single lorlatinib 100-mg dose in the presence of moderate CYP3A induction reduced plasma lorlatinib AUCinf and Cmax by approximately 23% and 22%, respectively, relative to a single lorlatinib 100-mg dose administered alone. In addition to CYP3A, lorlatinib metabolism is also mediated through UGT1A4, CYP2C8, CYP2C19, and UGT1A3. As modafinil does not induce the other metabolizing enzymes, this may explain the lower than expected 23% reduction in lorlatinib AUCinf following modafinil dosing. The AUCinf and Cmax values of M8, the primary metabolite of lorlatinib that is both formed and subsequently metabolized by CYP3A, decreased by approximately 40% and 35%, respectively, in the presence of moderate CYP3A induction. This decreased exposure may reflect the potentially greater effect of CYP3A induction on the metabolism of the M8 metabolite compared to its formation. Specifically, M8 formation likely occurs through many pathways in addition to CYP3A, whereas M8 elimination occurs more exclusively through CYP3A. This is also consistent with a similar reduction in M8 AUCinf noted in the prior drug interaction study with rifampin [6]. The absorption, distribution, metabolism, and excretion properties of lorlatinib have been characterized separately [10].
Changes in the estimated ratios for plasma 4 β-hydroxycholesterol/cholesterol and urine 6β-hydroxycortisol/cortisol have been shown to correlate with CYP3A activity in many prior clinical evaluations [11,12,13,14]. The two-fold increase in 4β-hydroxycholesterol/cholesterol ratios observed in this study is consistent with moderate CYP3A induction. However, the 6β-hydroxycortisol/cortisol ratio did not appear to increase over time. Possible explanations for these data include high inter-subject and intra-subject variability in the 6β-hydroxycortisol/cortisol measurements, making it a less reliable indicator of CYP3A activity than the 4β-hydroxycholesterol/cholesterol results [14, 15]; a small number of samples; and potentially higher stress-induced cortisol levels. Nearly all participants in this study experienced modafinil-related AEs, such as insomnia, anxiety, palpitations, and fatigue, and it is speculated that this may have increased participants’ stress, and correspondingly their cortisol levels after Period 2 day 1; this was reflected in the observed urine cortisol concentration (data not shown), which was noticeably higher on Period 2 days 8 and 15 when compared with Period 2 day 1. The higher stress-induced cortisol levels could have lowered the 6β-hydroxycortisol/cortisol ratios on days 8 and 15, thereby confounding the ability to see an overall change in the ratio.
A limitation of this study is that it included healthy participants who received a single dose of lorlatinib, rather than multiple doses. However, given the reduced lorlatinib plasma concentrations when co-administered with modafinil, it would not have been feasible or ethical to conduct this study in patients with non-small cell lung cancer, where reduced lorlatinib concentrations may have implications on reduced efficacy. Additionally, while the current study used modafinil, the magnitude of change in lorlatinib pharmacokinetics may be slightly different when co-administered with other moderate CYP3A inducers owing to variability in the extent of CYP3A induction and involvement in other enzyme pathways within this class of compounds. Finally, while data from six healthy participants convincingly demonstrated that it is safe to give lorlatinib 100 mg in combination with multiple-dose modafinil for the vast majority of patients, the current study was limited to a small sample size. Nevertheless, even if there exist outlier patients who, when administered the combination may experience transaminase elevations, these data strongly suggest that such AEs would be mild and not clinically meaningful as opposed to when lorlatinib is administered in combination with rifampin, and is therefore unlikely to require lorlatinib dose modifications.
5 Conclusions
Single-dose lorlatinib 100 mg was well tolerated when administered alone and in the presence of steady-state concentrations of the moderate CYP3A inducer modafinil, with no clinically meaningful AST or ALT elevations observed. Co-administration of lorlatinib 100 mg in the presence of steady-state concentrations of modafinil decreased the exposure to lorlatinib, reducing AUCinf and Cmax by 23% and 22%, respectively. This study supports the conclusion that lorlatinib 100 mg may be safely co-administered with moderate CYP3A inducers.
References
Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57(11):4720–44.
Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28(1):70–81.
Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci USA. 2015;112(11):3493–8.
Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–9.
Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 2018;19(12):1654–67.
Chen J, Xu H, Pawlak S, James LP, Peltz G, Lee K, et al. The effect of rifampin on the pharmacokinetics and safety of lorlatinib: results of a phase one, open-label, crossover study in healthy participants. Adv Ther. 2020;37(2):745–58.
Robertson P Jr, Hellriegel ET. Clinical pharmacokinetic profile of modafinil. Clin Pharmacokinet. 2003;42(2):123–37.
Robertson P Jr, Hellriegel ET, Arora S, Nelson M. Effect of modafinil on the pharmacokinetics of ethinyl estradiol and triazolam in healthy volunteers. Clin Pharmacol Ther. 2002;71(1):46–56.
Rowland A, van Dyk M, Warncken D, Mangoni AA, Sorich MJ, Rowland A. Evaluation of modafinil as a perpetrator of metabolic drug-drug interactions using a model informed cocktail reaction phenotyping trial protocol. Br J Clin Pharmacol. 2018;84(3):501–9.
Stypinski D, Fostvedt L, Lam JL, Vaz A, Johnson TR, Boerma JS, et al. Metabolism, excretion, and pharmacokinetics of lorlatinib (PF-06463922) and evaluation of the impact of radiolabel position and other factors on comparability of data across 2 ADME studies. J Clin Pharmacol. 2020;60(9):1254–67.
Wenk M, Todesco L, Krahenbuhl S. Effect of St John’s wort on the activities of CYP1A2, CYP3A4, CYP2D6, N-acetyltransferase 2, and xanthine oxidase in healthy males and females. Br J Clin Pharmacol. 2004;57(4):495–9.
Josephson F, Bertilsson L, Böttiger Y, Flamholc L, Gisslén M, Ormaasen V, et al. CYP3A induction and inhibition by different antiretroviral regimens reflected by changes in plasma 4beta-hydroxycholesterol levels. Eur J Clin Pharmacol. 2008;64(8):775–81.
Diczfalusy U, Nylén H, Elander P, Bertilsson L. 4β-hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br J Clin Pharmacol. 2011;71(2):183–9.
Dutreix C, Lorenzo S, Wang Y. Comparison of two endogenous biomarkers of CYP3A4 activity in a drug-drug interaction study between midostaurin and rifampicin. Eur J Clin Pharmacol. 2014;70(8):915–20.
Chen Y-C, Gotzkowsky SK, Nafziger AN, Kulawy RW, Rocci ML Jr, Bertino JS Jr, et al. Poor correlation between 6beta-hydroxycortisol:cortisol molar ratios and midazolam clearance as measure of hepatic CYP3A activity. Br J Clin Pharmacol. 2006;62(2):187–95.
Acknowledgements
The authors thank the study participants and the Brussels PCRU site personnel. The authors also thank Kimberly Lee of Pfizer Inc. for support with bioanalytical assays, Katherine Ginman and Michelle Bergeron for support with study oversight, Mochao Gao for support with programming, and Melissa O’Gorman for support with pharmacokinetic analysis for this study. Medical writing support was provided by Annette Smith, PhD, of CMC AFFINITY, McCann Health Medical Communications, and was funded by Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Pfizer Inc.
Conflict of interest
Jerry Li, Yazdi K. Pithavala, Jason Gong, Robert R. LaBadie, Josué Kunjom Mfopou, and Joseph Chen are employees of Pfizer Inc. and may own stock and/or stock options in Pfizer.
Ethics approval
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonisation Good Clinical Practice Guidelines. In addition, all local regulatory requirements were followed. Details of the independent ethics committee that approved the protocol are as follows: Comité d’Ethique Hospitalo-Facultaire Erasme-ULB, Route de Lennik 808, Brussels, BE-BRU B-1070, Belgium.
Consent to participate
All participants provided written informed consent before undergoing any study procedures.
Consent for publication
Not applicable.
Availability of data and material
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the USA and/or European Union or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Code availability
Not applicable.
Author contributions
JL study design, acquisition of data, interpretation of data. YKP study design, acquisition of data, interpretation of data. Jason Gong: study design, acquisition of data, interpretation of data. RRL study design, interpretation of data. JKM principal investigator of study, acquisition of data, interpretation of data. JC study design, acquisition of data, interpretation of data. All authors reviewed and provided intellectual contributions on previous versions of the manuscript, and read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Li, J., Pithavala, Y.K., Gong, J. et al. The Effect of Modafinil on the Safety and Pharmacokinetics of Lorlatinib: A Phase I Study in Healthy Participants. Clin Pharmacokinet 60, 1303–1312 (2021). https://doi.org/10.1007/s40262-021-01026-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01026-w