
Abstract
Huntington disease (HD) is a fatal progressive neurodegenerative disorder caused by an inherited mutation in the huntingtin (HTT) gene, which encodes mutant HTT protein. Though HD remains incurable, various preclinical studies have reported a favorable response to HTT suppression, emphasizing HTT lowering strategies as prospective disease-modifying treatments. Antisense oligonucleotides (ASOs) lower HTT by targeting transcripts and are well suited for treating neurodegenerative disorders as they distribute broadly throughout the central nervous system (CNS) and are freely taken up by neurons, glia, and ependymal cells. With the FDA approval of an ASO therapy for another disease of the CNS, spinal muscular atrophy, ASOs have become a particularly attractive therapeutic option for HD. However, two types of ASOs were recently assessed in human clinical trials for the treatment of HD, and both were halted early. In this review, we will explore the differences in chemistry, targeting, and specificity of these HTT ASOs as well as preliminary clinical findings and potential reasons for and implications of these halted trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Huntingtin (HTT)-lowering strategies are a promising avenue for the development of disease-modifying therapies for Huntington disease (HD), with antisense oligonucleotides (ASOs) being the first to reach clinical trials. |
The clinical trials of three HTT-lowering ASOs were recently halted; one due to worsening clinical outcomes, and the other two for a failure to lower mutant HTT. |
HTT, which is required for embryonic brain development, also has functions in the adult brain; it is unclear to what extent HTT lowering can be tolerated. |
Cerebrospinal fluid neurofilament light chain (NfL) emerges as a highly sensitive safety biomarker that may be predictive of unfavorable outcomes. |
1 Introduction
Huntington disease (HD) is an autosomal dominant neurodegenerative disorder caused by the expansion of the glutamine encoding CAG tract in exon one of the huntingtin (HTT) gene, which leads to the production of polyglutamine expanded mutant huntingtin (mtHTT) protein [1, 2]. HD manifests in people with 36 or more CAG repeats with full penetrance seen with 40 or more. The age of disease onset is influenced inversely by the length of the expansion, and symptoms do not typically develop until midlife [3, 4]. Symptoms of HD, including psychiatric disturbance, cognitive dysfunction, and the progressive loss of voluntary motor control, worsen over time until death, typically 15–20 years after motor onset [5].
There is no cure for HD and no disease-modifying treatments available. There is strong evidence that reduction of mtHTT could provide such a treatment. Conditional inactivation of mtHTT in adult, symptomatic HD mice results in the recovery of motor function and clearance of toxic protein aggregates [6]. In subsequent proof-of-concept studies, RNA interference was found to provide benefit to motor and neuropathological aspects of disease in multiple mouse models of HD [7,8,9], demonstrating the potential utility of suppression of mtHTT in adults as a disease-modifying therapy for HD.
One method of gene suppression is through antisense oligonucleotides (ASOs). ASOs are small single-stranded chemically modified DNA molecules that use Watson Crick base pairing to bind RNA and catalyze downstream events [10]. Gene suppressing ASOs form DNA/RNA heteroduplexes when bound, recruiting RNase H to degrade the targeted transcript. ASOs are a promising treatment option because they can bind to both mRNA and pre-mRNA, increasing the availability of targetable RNA sequences for selection. ASOs are also favorable as therapies for neurological diseases, such as HD, because they have long half-lives and broad distribution in the central nervous system (CNS), where they are taken up by neurons, glia, and ependymal cells without the assistance of a carrier [11].
Three ASO therapies for HD were recently assessed in human clinical trials (Table 1). Roche, in partnership with IONIS Pharmaceuticals, developed a non-selective total HTT-lowering ASO that progressed to a phase III trial, and Wave Life Sciences developed two allele-selective mutant HTT-lowering ASOs that were assessed in phase I/IIa trials. These ASOs, though all aimed at reducing the disease-causing protein, differ in a number of substantive ways that could affect their application, likely leading to the halting of all three of these trials for very different reasons. In this review, we provide an in-depth overview of the ASO therapies in recent halted HD human trials.
2 Chemistry
ASOs are synthetic, modified, single-stranded DNA molecules. Single-stranded DNA typically has a short half-life due to endonucleolytic degradation pathways so ASOs must be chemically modified to enhance stability in order to effectively reach their targets [12]. In most ASOs, this is accomplished by replacing the oxygen in the phosphodiester linkage with a sulfur to create a phosphorothioate (PS) linkage that is slightly more resistant to endonuclease activity [13]. Beyond stability, PS linkages enhance ASO distribution by forming disulfide bonds with albumin, the most abundant protein in blood plasma and the cerebrospinal fluid (CSF), which transports the ASO throughout the CNS [14, 15]. Aside from benefits to stability and distribution, PS linkages can cause immune activation. However, incorporating phosphodiester (PO) linkages into the sequence creating a mixed (PS/PO) backbone can minimize this response [16, 17]. Both the Roche/IONIS and Wave ASOs have (PS/PO) mixed backbones.
By chemically modifying the phosphodiesterase backbone of DNA to create PS linkages, stereochemistry is introduced, as each PS linkage becomes a chiral center. Each chiral center has the potential to yield a pair of enantiomers, 3D molecules with non-superimposable mirror image configurations denoted as Rp or Sp. Therefore, using the 2n rule to evaluate the permutability of the molecule, an ASO that is 18 base pairs in length and has 17 PS linkages (n), such as the FDA-approved spinal muscular atrophy (SMA) ASO, nusinersen, has 131,072 stereoisomeric forms that are given to patients as a racemic mixture [18]. PS/PO mixed backbones have fewer chiral centers and stereoisomers because PO linkages are achiral. Tominersen, the Roche/IONIS mixed backbone HD ASO, has 19 linkages, but since six linkages are achiral, there are only 8192 possible forms. Theoretically, each of the 8192 resulting molecules could have different pharmacologic properties. For instance, in ASOs, comparatively speaking, the Rp stereoisomer is expected to be favored by RNase H for cleavage while the Sp configuration offers increased stability [19,20,21,22]. Together the Rp and Sp linkages seem to create a functionally balanced molecule. Tominersen is stereorandom—a mixture of both Rp and Sp linkages.
Historically, the safety and potency of stereorandom drugs has been called into question. Although some stereoisomers may perform similarly therapeutic functions, there are others that are less than complementary, ranging from biologically inactive to highly toxic molecules [23, 24]. ASOs yield potentially hundreds of thousands of different stereoisomers making it technically prohibitive to use separation techniques to isolate one pure ASO molecule. In lieu of stereoisomer separation, Wave Life Sciences has developed stereocontrolled oligonucleotide synthesis with iterative capping and sulfurization (SOSICS) [22]. SOSICS grants control over the stereochemistry of each PS linkage during molecule production, allowing Wave to synthesize a single stereopure ASO molecule [25]. While this stereopure synthesis method eliminates the risk of enantiomeric impurities, controlling chirality influences drug activity [26]. Stereopure molecules composed of only Sp linkages demonstrate improved stability [22], but are significantly less potent [20]. RNase H has been reported to favor Rp linkages [19] but molecules composed of only Rp linkages do not show a significant increase in potency [20]. Controlling stereochemistry may provide enhanced stability, RNase H activity, or tolerability but the most potent ASO molecule may not be the most stable, potentially requiring a balance to be sought.
Regardless of stereochemistry, PS-modified ASOs have relatively low RNA binding affinity and are still substrates for endonucleases, limiting overall drug potency. ASO bases can be modified at the 2′ carbon of the sugar ring to enhance RNA binding affinity and improve stability [27]. Nucleosides containing these 2′ sugar modifications are not substrates for endonucleases so an ASO composed completely of 2′ modified sugars would not recruit RNase H. Therefore, ASOs that induce target degradation are designed as gapmers—chimeric oligonucleotides composed of the DNA sequence ‘gap’ that is susceptible to RNaseH-mediated degradation and the flanking chemically modified RNA wings, providing stability and affinity [28].The first 2′ sugar modification was the 2′-O-methyl (2′-O-Me), which increases the melting temperature of the molecule indicating improved stability. The Wave ASOs are 2′-O-Me gapmers, as the core sequence is flanked by 2′ sugar moieties with 2′-O-Me modifications and mixed PS/PO linkages [29]. Subsequent modifications, such as the 2′-O-methoxethyl (MOE) developed by IONIS Pharmaceuticals, have even higher RNA binding affinity and proved to be more stable than 2′-O-Me modifications [30]. The Roche/IONIS ASO is a MOE gapmer, as its core sequence is flanked by 2′ sugar moieties with MOE modifications and mixed PS/PO linkages [31].
3 Targeting
HD is an autosomal dominantly inherited disease, thus the causative mtHTT gene is the first logical therapeutic target. The HTT gene is haplosufficient, meaning that, while complete inactivation of HTT is embryonically lethal [33,34,35], the deletion of one copy of HTT does not cause an overt abnormal phenotype [36]. This suggests allele-selective lowering of mtHTT as a potential treatment for HD. This would typically be achieved by targeting the disease-causing mutation for suppression. However, the CAG tract that is expanded to cause HD is present in all HTT genes as well as many other genes throughout the genome [37]. Targeting the expanded CAG tract, therefore, has the potential for off-target hybridization—where the ASO binds to a similar sequence not within the gene of interest.
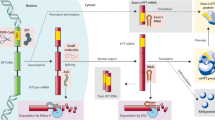
In lieu of targeting the mutation selectively, a nonselective approach was adopted to partially suppress both wild-type huntingtin (wtHTT) and mtHTT. In preclinical studies, 50% suppression of mtHTT is sufficient to provide benefit [38] and total HTT can be lowered by 50% without overt phenotype, [33], suggesting that appropriately dosed nonselective ASOs could provide a potentially safe and effective therapy. Nonselective ASOs, like the Roche/IONIS ASO, target HTT, indiscriminately binding to both wtHTT and mtHTT transcripts, and degrade a portion of each (Fig. 1).

Nonselective ASO-mediated suppression of total HTT. Figure by Erin Kenzie for Amber Southwell. Adapted from [44]. ASO antisense oligonucleotide, mRNA messenger ribonucleic acid, mtHTT mutant huntingtin, polyQ polyglutamine, RNaseH ribonuclease H, wtHTT wild-type huntingtin
While this nonselective strategy was widely pursued, population genetics studies identified single nucleotide polymorphisms (SNPs) that are in linkage disequilibrium with the CAG expansion (HD-SNPs), providing alternate targets for allele-selective mtHTT suppression [39, 40]. Human fibroblast lines were screened and selected for heterozygosity of HD-SNPs and subsequently used to screen potential allele-selective ASOs. This led to the development of a SNP-targeted ASO that discriminated and lowered fibroblastic mtHTT mRNA by a 5-fold difference compared with the wtHTT [41]. Single base-pair mismatches alter RNase H cleavage patterns, so ASOs were chemically modified to reduce RNase H activity outside the desired cleavage site, improving single nucleotide discrimination from 5-fold to 100-fold [42]. ASO selectivity was demonstrated in vivo using humanized HD mice that are heterozygous for human mtHTT and wtHTT transgenes, including some HD-SNPs [43], and the preclinical therapeutic benefit of this approach, including restoration of cognitive and behavioral deficits in previously impaired mice, has been established [44].
Targeting CAG expansion-associated SNPs, allele-selective HD ASOs can discriminate between the mtHTT and the wtHTT transcripts (Fig. 2) but SNP heterozygosity is required. Considering that haplotypes of upper and lower alleles, and thus heterozygosities, vary between individuals, this approach requires development of multiple ASO drugs targeting different SNPs in order to treat the majority of HD patients [45, 46]. Wave Life Sciences designed allele-selective ASOs that target two of these SNPs, rs362307 and rs362331. Based on HD population genetics, an estimated 36%–70% of HD patients globally are heterozygous for at least one of the two SNPs targeted by the allele-selective ASOs recently in clinical trials [32]. There is an estimated 8% of HD patients precluded from receiving SNP targeted allele-selective HD ASOs as they are homozygous for the known CAG expansion-associated SNPs [46] as well as a portion of individuals homozygous for the HD mutation for whom allele-selective HTT lowering via HD-SNP targeting would not be possible.

SNP-targeted allele-selective ASO-mediated suppression of mtHTT. Figure by Erin Kenzie for Amber Southwell. Adapted from [44]. ASO antisense oligonucleotide, mRNA messenger ribonucleic acid, mtHTT mutant huntingtin, polyQ polyglutamine, RNaseH ribonuclease H, SNP single nucleotide polymorphism, wtHTT wild-type huntingtin
4 Selectivity
Due to their differences in targeting, nonselective ASOs lower both wtHTT and mtHTT whereas allele-selective ASOs preferentially suppress mtHTT. While both approaches suppress mtHTT, the selective approach will leave more wtHTT production, potentially enhancing post-treatment ongoing HTT function. In adults, the extent of HTT function and its necessity is not fully understood.
It is known that HTT is required for normal brain development as ablation of the mouse HTT homolog (Hdh) in mouse embryos leads to reabsorption around embryonic day 8.5, the time when brain formation begins [33,34,35]. Genetically reducing HTT expression to ~ 33% of endogenous levels in embryos leads to mice born with misshapen heads and abnormal brain morphology [47]. Conversely, Hdh-het mice that are heterozygous for inactivation of HTT and express HTT at 50% have no overt phenotype, though on closer evaluation exhibit hyperactivity, some cognitive deficits, and neuronal loss in the subthalamic nucleus [34]. Similarly, there have been cases reported of adults who are heterozygous for HTT but still had normal neurodevelopment and function [48]. These data demonstrate that HTT is required for embryonic brain development, but that as little as 50% of the normal level is sufficient to perform this role. Interestingly, mice that only express mtHTT develop normally, suggesting that the HD mutation does not preclude this obligate role of HTT in brain development. Even in postnatal development HTT has obligatory roles as complete inactivation of (Hdh) in neonatal mice results in neurodegeneration and reduced survivability [49].
Further evaluating the effect of HTT deficiency, in a transgenic mouse model of post-development HTT loss, complete inactivation of Hdh in mice younger than 4 months caused acute pancreatitis whereas the inactivation of Hdh in mice older than 4 months had no impact on cell viability or motor function in one study [50]. Contrarily, in a similar study, complete inactivation of Hdh in adult mice led to widespread gliosis, thalamic calcification, and disturbed iron homeostasis [51]. Together these studies indicate post-development HTT lowering may be tolerated, though complete loss may have consequences at any age.
Since HTT is a key transcriptional regulator throughout development, it is perhaps unsurprising that, in adults, HTT has recently been implicated in corticospinal motor neuron repair, as these cells must first regress to an embryonic state to regenerate [52]. In neurogenesis, HTT is required for the proliferation and differentiation of neural stem cells [47], whereas mtHTT disrupts neuroepithelial junctions leading to premature differentiation [53]. During cell division, HTT is required for proper spindle placement in mitosis [54]. HTT becomes phosphorylated in response to elevated corticosterone levels, blocking neurogenesis [55]. In an HD mouse model, cell proliferation is reduced, suggesting that mtHTT may not be as effective as wtHTT in facilitating neurogenesis [56]. Supporting this is the finding that adult striatal neurogenesis is diminished in the HD brain [57].
After development and neurogenesis, one of the ongoing roles of HTT is as a scaffold protein in a variety of cellular processes. HTT colocalizes with postsynaptic density-95 (PSD-95) on postsynaptic striatal projection neurons (SPNs) [58]. HTT protein interacts with huntingtin associated protein 1 (HAP1) to recruit dynactin to the vesicles, inducing axonal transport of neurotrophic factors like brain-derived neurotrophic factor (BDNF) [59]. To provide the ATP necessary for fast axonal transport, HTT binds the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) enzyme to catalyze vesicular glycolysis [60]. Complexing with huntingtin associated protein 40 (HAP40) and Rab5, HTT modulates motility in early endosomes [61]. HTT facilitates selective autophagy by recruiting proteins required for cargo loading [62, 63]. Involved in the transport of autophagosomes along the neuronal process, a decrease in available HTT leads to reduced motility and incomplete degradation of cargo [64]. Likewise, in mitophagy, which is the selective degradation of dysfunctional mitochondria, HTT acts as a scaffold [65, 66]. Mitochondria produce reactive oxygen species (ROS) as a by-product of metabolism, but as dysfunctional mitochondria produce ROS in excessive quantities, impaired mitophagy leads to increased oxidative stress, which is seen in HD brains [67].
HTT is also involved in ongoing stress signaling in neurons. HTT localizes to the endoplasmic reticulum (ER) but can travel to the nucleus to signal ER stress [68]. HTT mitigates the oxidative stress response through promotion of NRF2 activity [69, 70]. In response to damage from oxidative stress, HTT localizes to the site of DNA damage and recruits DNA repair proteins [71]. Accumulation of mtHTT impairs this stress response and makes neurons more sensitive to ROS [72]. The combined presence of mtHTT and endotoxins causes microglia to release more inflammatory cytokines [73]. Expression of mtHTT makes striatal cells susceptible to loss of BDNF leading to glutamate excitotoxicity [74]. HTT is neuroprotective and can prevent apoptosis in response to excitotoxicity [75]. As part of the canonical stress response, HTT frees up ATP by localizing to cofilin to prevent actin remodeling [76] and through the formation of huntingtin stress bodies (HSBs), which halt endosomal trafficking [77]. In the body, prolonged stress may interfere with HTT function. Moreover, mtHTT fails to adequately perform many stress response roles, leading to increased basal cellular stress in the HD brain.
As HTT serves such a critical role in the development and maintenance of the nervous system, the complete loss of available HTT protein at any stage of life would likely be detrimental. Partial suppression in adults appears tolerated [38, 78,79,80], though the threshold as to safe HTT loss is unclear. The interpretation of results from these studies may be limited, as HTT lowering was only evaluated for 6–9 months in healthy animals in a controlled lab environment.
These studies were conducted using animal models, most not HD models. In healthy adult mice and monkeys that lack mtHTT, lowering of total HTT is well tolerated. However, in mouse models of HD, phenotypes vary depending on mtHTT expression level. The zQ175 knock-in model of HD is hypomorphic, meaning that knock-in mtHTT expression is very low. When this transgene is expressed in FVB mice (Q175F), mice display early death from fatal seizures. These mice express 50% wtHTT and a small amount of mtHTT. Yet, Hdh-het mice on the FVB strain, which also have 50% wtHTT but lack mtHTT, do not display early fatal seizures, though they may still develop spontaneous seizures later in life at a proportion greater than that seen in WT FVB mice. Thus, the small amount of mtHTT transforms the late onset, moderate seizure phenotype with low penetrance to an early onset, fatal phenotype with higher penetrance. Importantly, this is not an aspect of HD. In this case, the addition of a small amount of mtHTT exacerbates an existing HTT deficiency phenotype of Hdh-het FVB mice. Q175FDN mice have the same knock-in as Q175F mice but with restored mtHTT expression. Q175FDN het mice have 50% wtHTT and increased mtHTT expression yielding total HTT levels closer to WT mice. The increased mtHTT in these mice protects them from the early fatal seizures, transforming it back to a late onset, low penetrance seizure phenotype similar to Hdh-het mice. However, these mice do display reduced survival compared with WT mice following an extended period of HD-like neurological decline. Thus, they die from HD, unlike the Q175F mice with reduced total HTT and a small amount of mtHTT that die from mtHTT-exacerbated HTT deficiency [81]. This demonstrates that the threshold of tolerability for HTT loss may be very different in HD patients than in the healthy mice and monkeys used to assess preclinical tolerability of HTT suppression.
The question of whether HTT reduction is tolerated is by no means black and white, so it is necessary to weigh the costs versus benefits of HTT lowering. In humans, like the Hdh-het mice, who are heterozygous for reduced wtHTT but lack mtHTT, neurodevelopment and brain function are phenotypically normal [48]. Yet, just as the increase in mtHTT protein in the Q175FDN mice protected from loss of HTT effects, it also led to more severe HD-like pathology. There is also evidence, in humans, that mtHTT is more toxic than loss of HTT. There is a SNP in the NF-kB binding site in the HTT promoter that inhibits binding, resulting in modest downregulation of HTT. When found on the wtHTT allele, modestly reducing wtHTT expression throughout life, it accelerates age of HD onset on average ~ 4 years. Conversely, when the SNP is on the mtHTT allele, modestly reducing mtHTT expression throughout life, disease onset is delayed by ~ 9 years [82].This is further supported by a study in humanized HD mice showing that nonselective HTT lowering dose-dependently increases striatal volume of HD mice, ameliorating atrophy, but the opposite effect was observed in non-HD mice, with a dose-dependent reduction in striatal volume [83].
Taken together, these data suggest that mtHTT is more toxic than loss of total HTT, and in a nonselective approach, the benefit of lowering mtHTT should outweigh any potential detriment from loss of total HTT. However, it is likely that below some threshold of total HTT loss, the detriment may reduce or outweigh benefit.
5 Summary of Clinical Findings
5.1 Tominersen
Ionis Pharmaceuticals, in partnership with Roche, developed tominersen, a nonselective stereorandom 2′ MOE gapmer ASO that has completed a phase I/IIa clinical trial, an open-label extension study, and a phase III clinical trial.
5.1.1 Phase I/IIa Clinical Trial
In this trial, participants received intrathecal injections of 10, 30, 60, 90, or 120 mg of the ASO or placebo every 4 weeks for a duration of 16 weeks. The primary endpoint of this trial was safety, assessed in part by tolerability. Of the 46 participants enrolled, all completed the trial. In both the treatment and placebo groups, 98% of participants reported adverse events, and of those, 83% were mild with 6% considered to be drug related, none of which were serious adverse events. Secondary endpoints included quantitation of CSF mtHTT protein as a potential measure of target engagement, CSF neurofilament light chain (NfL) and Tau as potential measures of neurodegeneration, and neuroimaging. CSF samples were collected and assessed longitudinally at each injection as well as the two post-dosing 4-week time points at days 113 and 141. In participants receiving tominersen, there was an observed dose-dependent lowering of mtHTT in the CSF. An average mtHTT reduction of 20–42% was recorded, with a maximum reported lowering of 63%, suggesting that HTT protein was successfully lowered in the CNS. The CSF concentrations of NfL and Tau in participants that received injections of tominersen 90–120 mg increased 1–2 months after the final injection. These biomarkers returned to baseline after a reported 7–27 months following the cessation of treatment. While NfL has now emerged as a potential biomarker of clinical significance, at the time of this trial, and in the context of the otherwise satisfactory trial outcomes, the clinical implications of this finding were unclear. In hindsight, this may have been an early indication that chronic dosing would lead to poor tolerability over time. Furthermore, there were reported increases in ventricular volume at days 113 and 197 in the patients from the treatment group that were not associated with any known adverse events. This was seen across the ASO dosing groups, and though the 120-mg dosage group showed the greatest increase in volume, the 10-mg and 90-mg dosage groups demonstrated a similar degree of enlargement suggesting that this was not fully dose dependent.
As intracellular proteins, the release of NfL and Tau into the CSF could be the result of axonal loss, remodeling, or toxicity, which may be in part due to the on-target effects of HTT lowering or potential off-target liabilities, such as aptameric effects or the activation of immune pathways. For example, the aptameric or protein oligonucleotide interactions between the PS linkages of ASOs and albumin facilitates transport and distribution throughout the CNS [14, 15]. However, PS linkages can interact adversely with other proteins and can cause immune activation. The half-life of ASOs in the brain is over 100 days [84], meaning that administration every 4 weeks could result in a buildup of ASO in the brain, potentially exceeding planned dosages. In this trial, it is important to note that while there was no reported ASO build up in the plasma or the CSF [31], CSF mtHTT levels were trending down [85], which may have led to HTT lowering that exceeded the therapeutic target range.
5.1.2 Open-Label Extension Study
At the completion of the phase I/IIa trial [31, 85], dosing was discontinued in all patients prior to initiation of the open-label extension (OLE), which was designed to evaluate the safety and tolerability of tominersen 120 mg at different dosing intervals. Following an interval of 7–27 months after final dosing in the phase I/IIa study, during which CSF mtHTT and NfL levels returned to baseline, participants were randomized to either the 4-week or 8-week dosing arms. All participants received two 120-mg loading doses of ASO administered intrathecally, 4 weeks apart. After this, patients received doses according to their assigned dosing arm. Patients in the 4-week arm continued receiving 120 mg of the ASO every 4 weeks, while patients in the 8-week arm began receiving 120-mg injections of ASO every 8 weeks. Thus, following the loading doses, the 4-week dosing group received an additional 14 doses, while the 8-week dosing group received an additional seven.
Over the course of 15 months, there were a total of 621 adverse events reported. The 4-week group accounted for 412 of these adverse events and, of the 23 patients in this group, 100% experienced at least one adverse event. There were a total of 14 serious adverse events that were reported in five of the 23 patients. Of these 14 serious adverse events, there were seven that were suspected to be drug related and they occurred in only two of the five patients. Meanwhile, 98% of the 23 patients in the 8-week group reported at least one adverse event, accounting for 209 of the 621 total adverse events. There were six serious adverse events reported in three patients in this group, none of which were suspected to be drug related.
In each group, CSF mtHTT reduction and NfL levels were monitored over the duration of the trial. In the 4-week dosing group, CSF mtHTT lowering exceeded the therapeutic target range of 30–50% with a mean 70% trough reduction. The 8-week dosing group remained in therapeutic target range with a mean 44% trough reduction. There were frequent transient elevations in the CSF white blood cell counts of patients in the 4-week group that were not linked to any known adverse events. In the 8-week group, elevations were minimal and infrequent. Similarly, there were elevations in CSF protein in the 4-week group, reaching an apparent maximum of 500 g/L compared with the 8-week group, which had roughly < 250 g/L. On day 141, in the 4-week group, there was a spike in CSF NfL concentrations that mirrored the phase I/IIa in both timing and magnitude and remained 53% above baseline at the end of the 15-month study. Although there was still an increase in NfL in the 8-week arm at the same time as in the 4-week arm, it was of a lower magnitude that did not reach statistical significance and remained only 10% above baseline at the 15-month timepoint. Moreover, the 8-week arm achieved steady-state CSF exposure at roughly 1.6 ng/mL, whereas the 4-week arm showed a steady rise through the first 12 months of treatment, reaching a peak exposure of roughly 5.8 ng/mL. Both the 4-week and 8-week arms had similar trough exposure in plasma [85].
Through less frequent dosing, the study demonstrated steady-state exposure and some mitigation of the transient CSF NfL elevations. Using the data from this trial and the phase I/IIa trial, a pharmacokinetic/pharmacodynamic (PK/PD) model was developed which showed that 8-week dosing was predicted to exceed the preclinical efficacy threshold in the cortex and the caudate, whereas a 16-week dosing arm was only predicted to exceed the efficacy threshold in the cortex. Together these findings supported revision of the dosing schedule for subsequent trials.
5.1.3 Phase III Clinical Trial
Tominersen was assessed in a phase III pivotal trial evaluating the therapeutic efficacy and safety of 120 mg delivered at either 8- or 16-week dosing intervals. At the initiation of the trial, two 120-mg loading doses of the ASO were intrathecally administered to every ASO treatment patient 4 weeks apart in an effort to reach and maintain steady-state drug concentrations before continuing the experimental dosage schedule of every 8 weeks [86]. One group of ASO-treated participants received the drug at every injection, while another group received alternating 120-mg injections of ASO and placebo, resulting in an 8-week treatment group and a 16-week treatment group. To evaluate the efficacy of the ASO, each participant in this trial received a composite Unified Huntington Disease Rating scale (cUHDRS) score and a Total Functional Capacity (TFC) score, assessing for everyday capabilities.
In March of 2021, an independent safety monitoring board found that the risk/benefit ratio to patients did not support further dosing [87]. The preliminary data from this trial revealed poor safety signals. Of the 260 patients in the 8-week dosing group, 90% reported adverse events and 15% reported a serious adverse event. In the 16-week dosing group, 86.2% of the 261 patients experienced an adverse event while 8.4% experienced serious adverse events. Lastly, the placebo group was made up of 260 patients, 90.4% of whom had an adverse event and 10.8% had a serious adverse event. Overall, the 8-week dosing group had the highest number of adverse events and serious adverse events, while the 16-week dosing group had the fewest, suggesting that the 8-week dosing was not as well tolerated as the 16-week dosing or placebo groups, which were comparable. While fluid biomarker data is still pending, CSF mtHTT reduction is expected to mirror previous trials, yet the preliminary findings from this study indicate that there may be no associated therapeutic benefit [86]. There is little evidence that the treatment halted or reversed disease progression and there are indications that the treatment may have actually exacerbated it over time. Total Motor Score (TMS), Stroop Word Reading Test (SWR), and Montreal Cognitive Assessment (MoCA) scores were comparable between the 16-week dosing group and the placebo group, but in the 8-week group, the scores were diminished, indicating an association between the treatment and the deterioration of cognitive and motor capabilities. Moreover, in the span of 69 weeks there was an observed dosing frequency-dependent decline in cUHDRS, TFC, and Clinical Global Impression of Severity (CGI-S) scores, suggesting that overall clinical performance in patients receiving tominersen worsened more so than it did in patients with untreated disease. Still, both the trial and the OLE have continued without dosing, allowing for further analysis.
Considering the dosing frequency effect observed with the 8-week dosing group performing worse than the 16-week dosing group, ongoing studies to compare ASO exposure and target engagement to specific clinical measures may provide additional insight into the risk/benefit profile of this treatment. One such insight comes from the observed changes in ventricular volume over the course of the treatment. While the phase I/IIa clinical trial showed an increase in ventricular volume that was not wholly dose-dependent [31], the phase III clinical trial revealed an observed dose frequency-dependent response. There was an 11.4% increase in the ventricular volume of the placebo group, whereas the 8-week and 16-week group had a ventricular volume increase of 24.3% and 17.3%, respectively. This was one area in which the 16-week dosing group was not similar to the placebo. Still, the volumetric analyses of the whole brain and the caudate showed that, after 15 months, there were no drug-related changes in the 8- and 16-week dosing groups when compared with placebo. As there were no significant changes in caudate volume between the ASO treatment groups when compared with placebo, the increase in ventricular volume is more likely due to increased CSF volume, rather than atrophy, which may indicate inflammation.
5.2 Wave Life Sciences WVE-120101 and WVE-120102
Wave Life Sciences developed two stereopure allele-selective ASOs, WVE-120101 and WVE-120102, each targeting a different CAG-associated SNP, rs362307 (SNP1) or rs362331 (SNP2). Each SNP is heterozygous in ~40% of HD patients of European descent [45, 46]. Since some patients are heterozygous for more than one of the two targeted SNPs, it is estimated that in combination, these two allele-selective ASOs could potentially offer therapeutic coverage to 36%–70% of the HD population [45]. Recently, both of these allele-selective ASOs underwent phase I/Ia clinical trials, PRECISION-HD1 and PRECISION-HD2, evaluating the safety and tolerability of the drugs in dosages of 2, 4, 8 16, and 32 mg. Early topline data for PRECISION-HD2, including dosing of up to 16 mg, was released in December of 2019. At these dosages, 72% of the 31 participants in the treatment group and 83% of the 13 participants from the placebo group experienced an adverse event, none of which were considered serious [88]. Meanwhile, in the PRECISION-HD1 study, 91% of the patients in the treatment group and 75% of those in the placebo group reported an adverse event, though none were considered serious [89]. In participants receiving WVE-120102, there was a statistically significant mean reduction of 12.4% in CSF mtHTT across dosage groups and no changes in CSF NfL were observed [88]. In participants receiving WVE-120101, CSF mtHTT reduction trended downward but, when compared with the placebo group, did not reach statistical significance [89]. Though these seemed to be modest reductions compared with the nonselective HD ASO, the maximum dosages administered in these trials was 16 mg, whereas the tominersen trials included dosages of up to120 mg.
A 32-mg treatment arm was initiated to evaluate whether dose escalation could safely lead to an effective mtHTT knockdown. The PRECISION-HD2 32-mg arm began dosing in January 2020 [90] and the PRECISION-HD1 32-mg arm, which was delayed due to COVID-19 [91], began dosing in March 2020 [90]. However, based on the results from PRECISION-HD2, both studies were halted in early 2021 due to a failure to consistently lower mtHTT in patient CSF [89, 92]. While the PRECISION-HD1 data is still pending, the data from the PRECISION-HD2 32-mg treatment group shows that, despite increasing the dosages of the respective ASOs, there were no significant reductions in CSF mtHTT over time. The mean CSF mtHTT reduction did not vary between treatment groups; the lack of a dose response further indicating that the drug had failed to engage the therapeutic target. In terms of overall tolerability of WVE-120102, 83% of the 66 participants in the treatment group and 90% of the 22 patients in the placebo group reported adverse events. In the 32-mg treatment group alone, seven of the 13 patients reported a serious adverse event, such as amnesia, ataxia, delirium, disorientation, and slurred speech. These serious adverse events were transient, but they led six of the 13 patients in the 32-mg group to discontinue treatment. NfL did not increase over time and though cUHDRS scores did not improve, they were not significantly worse than the natural history study projections suggesting that the ASO treatment was not directly associated with the exacerbation of HD progression. However, increasing the dose to 32 mg did not lead to a significant knockdown in CSF mtHTT but instead raised concerns about the underlying safety and tolerability of the platform. At this time, Wave does not plan to continue development of WVE-120101 and WVE-120102 but will instead focus on a different allele-selective HD ASO [92].
In November 2020 [93], Wave Life Sciences announced the development of WVE-003, an allele-selective ASO targeting a third HD-SNP that is targetable in an estimated 40% of the HD patient population [89]. The WVE-003 ASO reportedly has increased potency, distribution, and a longer half-life in the CNS due to the incorporation of novel backbone chemistry. Wave has introduced phosphoramidate diester (PN) backbone modifications by substituting phosphoryl guanidine for sulfur or oxygen onto existing PS/PO backbones, creating PS/PO/PN backbones. While PS/PO backbones are negatively charged, PN modifications are uncharged, likely contributing to enhanced distribution. Based on favorable pre-clinical mtHTT-lowering data in motor neurons and BACHD mice [92], Wave is moving forward with WVE-003 and expects to begin a phase I trial sometime in 2021.
6 Concluding Remarks
The ASOs discussed here are the first of the many HTT-lowering therapies in development to reach clinical trials. ASOs are useful for this purpose as they distribute widely throughout the CNS and do not require a viral or lipid carrier. In non-human primate models, single-stranded ASOs administered directly to the CSF are distributed widely throughout the cortical regions of the brain [44, 94] that are involved in cognition, but are not as effective as viral therapies in reaching subcortical structures, such as the basal ganglia and limbic system, which influence motor function and psychiatric symptoms in HD [95]. With the approval of nusinersen for SMA, ASOs also have an established track record for successful and tolerated CNS application.
This year the clinical trials for three HD ASOs were halted. These trials were conducted in early-manifest HD patients. Roche/Ionis required a cUHDRS independence scale score of 70/100 and Wave required a total functional capacity score between 7 and 13, excluding later-stage patients that have declined to the point of needing assistance in self-care. Both trials stipulated cUHDRS diagnostic confidence scores of 4, meaning there was 99% confidence that there was motor-manifest HD, excluding prodromal participants. HD therapies may have less of an effect on late-stage HD because there is greater damage accumulated, while prodromal participants may not demonstrate measurable progression during the course of the trial.
If therapeutic efficacy is demonstrated in early-manifest individuals, future studies should include both the later-stage HD and prodromal participants to assess whether HTT ASOs can effectively reverse, delay, or even prevent disease onset. Though the possibility of preventative treatment options is appealing, it raises the question of when is the ideal time to begin therapeutic intervention in people who are still healthy. The psychiatric symptoms of HD can appear up to 12 years before emergence of motor dysfunction [96]. Cognitive decline can begin an estimated 15 years before motor HD manifests [97]. Neuronal damage can begin at least 24 years prior to predicted disease onset as elevated mtHTT and NfL are detected in the CSF of young gene carriers [98]. However, HTT-lowering intervention prior to age 26 years should be approached cautiously considering that HTT is critical for brain development, which is not complete until age 25 [99]. This presents a challenge for the treatment of juvenile-onset HD as symptoms manifest during post-natal brain development.
The nonselective approach is attractive as it offers a universal treatment option to the majority of the HD community, but the tolerability of such an approach has been called into question. The findings from the most recent trial indicate that tominersen, at least at 120 mg, is not well tolerated [86], yet the cause of this remains unclear. Based on the transient increase in CSF NfL levels that was seen at the same post-treatment interval in the OLE, in the 4-week and, to a lesser magnitude, the 8-week dosing groups, dosing frequency likely played a role in this negative outcome. Taking into account the aggressive dosing strategy and the impressive half-life of ASO drugs in the brain, tominersen may have been better tolerated in smaller doses. Still, it remains to be seen whether poor tolerability was driven by off-target aptameric or inflammatory effects or by on-target HTT loss.
Considering that ventricular enlargement was not accompanied by caudate volume loss, suggesting hydrocephalus rather than accelerated atrophy, this may be a non-specific effect of ASO chemistry. This is further evidenced by the transient increase in the CSF NfL. CSF NfL is currently being studied as a marker in neurodegenerative diseases [100,101,102] and pre-clinical animal models of HD [103]. NfL has also been found in higher concentrations in plasma and CSF of HD and pre-HD patients and is thought to be related to neuronal cell death [104]. However, since acute neurotoxicity leads to transient increases in CSF mtHTT [105, 106], which is known to predominantly originate from neural tissues of the CNS [105], the concomitant reduction in CSF mtHTT suggests that the increase in CSF NfL may not necessarily be tied to cell death. Considering that HTT is mostly perinuclear, while NfL is found throughout processes, both would be released by cell death, while NfL could be preferentially released by changes affecting cell processes, such as de-arborization and loss of connectivity. Changes to neuronal processes are important in neurodevelopment but also occur throughout life due to toxicity and disease and are thought to be responsible for age-related increases in CSF NfL [107]. In other neurodegenerative diseases, such as multiple sclerosis and spinal muscular atrophy, reductions in CSF NfL concentrations are associated with improved clinical outcomes [108], suggesting that this may also be an important marker of therapeutic efficacy in HD. Overall, the tominersen program showed that increased CSF NfL may be predictive of worsening clinical outcomes. Despite the transience of the observed spikes, NfL appears to be a highly sensitive safety signal and should be closely monitored in future clinical trials for HD. Going forward, even transient changes should be considered an early indicator of reduced tolerability.
Aside from the transient increases in CSF NfL, elevations in CSF white blood cell counts and protein concentrations [85] may further support the onset of acute neuroinflammation. While extensive preclinical safety in non-human primates was performed prior to human trials, human-specific liabilities cannot be ruled out. Additionally, preclinical safety was performed in healthy monkeys, and considering the increased sensitivity of HD neurons to any type of stress, it is possible that the HD neurons were unable to tolerate the ASO-induced effects as well as healthy neurons. In an in vivo evaluation of pre-trial ASOs, an allele-selective ASO passed the initial tolerability screens, including acute tolerability in HD mice as well as repeated dosing and overdosing in wild-type rats and mice [43]. However, in a longer-term preclinical efficacy study, the same ASO induced hind limb ataxia and death in multiple HD models, regardless of whether the target sequence was present [44]. Thus, this ASO drug that was well tolerated in healthy brains was toxic in HD brains independent of HTT-lowering activity. This suggests that HD brains are more susceptible to non-specific ASO toxicity, and that some tolerability liabilities may not be uncovered by preclinical safety in healthy animals. This is, in fact, quite similar to what occurred with tominersen, which demonstrated excellent preclinical safety in healthy animals as well as subacute tolerability in HD patient brains during phase I testing. Overt toxicity was not apparent until long-term delivery of high doses to people with HD. It would be interesting to determine whether testing in HD non-human primates could yield more stringent preclinical safety standards and could have detected reduced tolerability of tominersen. However, we lack robust primate models of HD, reducing feasibility of this approach. The toxicity of the drug may explain why, in the phase III clinical trial, lowering CSF mtHTT failed to offer therapeutic benefit [86], as it seems probable that the poor tolerability in these highly susceptible HD neurons outweighed and likely masked any benefit from the suppression of mtHTT.
On-target toxicity related to total HTT loss is also a possibility. Though suppression of mtHTT is beneficial and partial suppression of wtHTT has been well tolerated in animals, it is unknown to what extent wtHTT can be safely lowered in the HD brain. Throughout the course of the trial, CSF mtHTT was used as a marker for target engagement and while this data has yet to be released, the PK/PD model, which is based on data from preclinical non-human primate studies as well the data from on the phase I/IIa clinical trial and OLE supports a target therapeutic range of 30–50% CSF mtHTT reduction [85]. Due to the size and complexity of the human brain, ASO distribution is uneven and may potentially yield high magnitude HTT reduction in regions that are in close contact with CSF. Using the data from a study in which the endogenous wild-type HTT was knocked down in the brains of non-human primates [38], it was predicted that a CSF mtHTT knockdown of 30–50% could potentially indicate a cortical mtHTT reduction of 55%–80% and a caudate mtHTT reduction of 25–45% [85]. While data from primates is clearly more similar to humans than the smaller and simpler rodent brains, it is important to keep in mind that a monkey brain is still much smaller and less complex than a human brain, and a monkey spinal column much shorter than that of an adult human. Thus, it is unclear to what extent this data is predictive of ASO distribution and activity in the HD patient brain.
It is also unclear whether or not the alternative splice variant of HTT exon 1 [109, 110] would be suppressed by this ASO. Because ASOs are active against pre-mRNA, in the nucleus of cells, they are able to induce degradation of transcripts prior to splicing events, so could reduce both the full-length and exon 1 splice variants of HTT. However, the target of tominersen, in exon 36, is distal to the exon 1 transcript, and depending on kinetics of RNA processing, this splice variant may still be generated, potentially leading to ongoing pathogenesis in the presence of full-length HTT suppression. However, evidence from mice suggests that ASO targeting outside of exon 1 is therapeutically efficacious. Additionally, there are two modifications of the YAC128 HD model, the C6R line [111] and the shortstop line [112], that express identical exon 1 regions to the symptomatic YAC128 line, but have alterations downstream in the gene that mitigate toxicity and prevent neurodegeneration. This suggests that, at least in mice, a distinct exon 1 transcript is not the major driver of disease. Nonetheless, considering the validated CSF mtHTT lowering by tominersen, the question remains whether the lack of therapeutic efficacy is related to non-specific ASO toxicity that masks any benefit in sensitive HD neurons, excessive total HTT knockdown in superficial brain regions resulting in HTT deficiency toxicity, inefficient knock-down of HTT exon 1 transcripts, or insufficient mtHTT knockdown in critical deep brain regions, such as the caudate nucleus.
For this reason, the allele-selective approach, though more technically challenging with each drug only applicable to a portion of the HD population, may be more effective in the long run because it provides the benefit of mtHTT lowering without the potential detriment of wtHTT loss. Yet, the most recent trials were halted due to the apparent absence of a dose-dependent response, failure to consistently lower mtHTT in patient CSF, and poor tolerability at higher doses. There were no reported increases in CSF white blood cells or proteins suggesting that the toxicity may not be neuroinflammatory in nature, but the exact cause remains unclear. Regardless, at 32 mg the drug reached the maximum tolerated dosage and still did not induce significant mtHTT knockdown. These ASOs were advanced to clinical trials in the absence of preclinical therapeutic efficacy or tolerability studies in HD models, which may have been able to determine that the effective dose was greater than the tolerated dose. While there are no mouse models of HD that include SNP1, preclinical tolerability studies in HD mice may have provided useful information. Additionally, the Hu97/18 model developed in 2013 [113] is heterozygous for SNP2 with the SNP on the mutant allele, thus could have been used for both preclinical tolerability and efficacy assessment.
The failure of these trials highlights the need for preclinical evaluation in appropriate models of disease. It is not enough to demonstrate target engagement in patient cells and tolerability in healthy animals. Ideally, preclinical assessments of therapeutic efficacy should be performed but, at the minimum, long-term tolerability in rodent models of HD should be assessed prior to clinical translation of all experimental HD therapies. However, there are currently no regulatory requirements for preclinical safety testing in diseased animals, so these studies are not always performed.
While all three recent HTT-lowering ASO trials failed, they did so for very different reasons. Despite these outcomes, it is still our belief that HTT is a promising therapeutic target and both ASO strategies will be necessary to meet the needs of the HD community for disease-modifying therapy.
References
De Rooij KE, De Koning Gans PA, Roos RA, Van Ommen GJ, Den Dunnen JT. Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum Genet. 1995;95(3):270–4. https://doi.org/10.1007/bf00225192.
Young AB. Huntingtin in health and disease. J Clin Invest. 2003;111(3):299–302. https://doi.org/10.1172/JCI17742.
Djoussé L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am J Med Genet A. 2003;119A(3):279–82. https://doi.org/10.1002/ajmg.a.20190.
Lee J-M, Correia K, Loupe J, Kim K-H, Barker D, Hong EP, et al. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. 2019;178(4):887-900.e14. https://doi.org/10.1016/j.cell.2019.06.036.
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98.
Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101(1):57–66. https://doi.org/10.1016/s0092-8674(00)80623-6.
Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, et al. From the cover: RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. PNAS. 2005;102(16):5820–5. https://doi.org/10.1073/pnas.0501507102.
Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol Ther. 2005;12(4):618–33.
Wang Y-L, Liu W, Wada E, Murata M, Wada K, Kanazawa I. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci Res. 2005;53(3):241–9.
Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–93. https://doi.org/10.1146/annurev.pharmtox.010909.105654.
Southwell AL, Skotte NH, Bennett CF, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18(11):634–43. https://doi.org/10.1016/j.molmed.2012.09.001.
Wu Y, Lu J, Kang T. Human single-stranded DNA binding proteins: guardians of genome stability. Acta Biochim Biophys Sin. 2016;48(7):671–7. https://doi.org/10.1093/abbs/gmw044.
Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46(4):1584–600. https://doi.org/10.1093/nar/gkx1239.
Crooke ST, Vickers TA, Liang X-H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020;48(10):5235–53. https://doi.org/10.1093/nar/gkaa299.
LeVine SM. Albumin and multiple sclerosis. BMC Neurol. 2016;16:47. https://doi.org/10.1186/s12883-016-0564-9.
Zhou W, Agrawal S. Mixed-backbone oligonucleotides as second-generation antisense agents with reduced phosphorothioate-related side effects. Bioorg Med Chem Lett. 1998;8(22):3269–74. https://doi.org/10.1016/S0960-894X(98)00591-5.
Pisetsky DS, Reich CF. Influence of backbone chemistry on immune activation by synthetic oligonucleotides. Biochem Pharmacol. 1999;58(12):1981–8. https://doi.org/10.1016/S0006-2952(99)00294-4.
(TGA) TTGA. Australian Public Assessment Report for nusinersen (as heptadecasodium). Commonwealth of Australia: Australian Government Department of Health Therapeutic Goods Administration (TGA); 2018.
Koziolkiewicz M, Krakowiak A, Kwinkowski M, Boczkowska M, Stec WJ. Stereodifferentiation—the effect of P chirality of oligo (nucleoside phosphorothioates) on the activity of bacterial RNase H. Nucleic Acids Res. 1995;23(24):5000–5. https://doi.org/10.1093/nar/23.24.5000.
Østergaard ME, De Hoyos CL, Wan WB, Shen W, Low A, Berdeja A, et al. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020;48(4):1691–700. https://doi.org/10.1093/nar/gkaa031.
Yu D, Kandimalla ER, Roskey A, Zhao Q, Chen L, Chen J, et al. Stereo-enriched phosphorothioate oligodeoxynucleotides: synthesis, biophysical and biological properties. Bioorg Med Chem. 2000;8(1):275–84. https://doi.org/10.1016/S0968-0896(99)00275-8.
Iwamoto N, Butler DCD, Svrzikapa N, Mohapatra S, Zlatev I, Sah DWY, et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat Biotechnol. 2017;35(9):845–51. https://doi.org/10.1038/nbt.3948.
Hartigan-Go K, Bateman DN, Daly AK, Thomas SHL. Stereoselective cardiotoxic effects of terodiline. Clin Pharmacol Ther. 1996;60(1):89–98. https://doi.org/10.1016/S0009-9236(96)90171-X.
Scott AK. Stereoisomers and drug toxicity. The value of single stereoisomer therapy. Drug Saf. 1993;8(2):149–59. https://doi.org/10.2165/00002018-199308020-00005.
Oka N, Wada T. Stereocontrolled synthesis of oligonucleotide analogs containing chiral internucleotidic phosphorus atoms. Chem Soc Rev. 2011;40(12):5829–43. https://doi.org/10.1039/c1cs15102a.
Huang K, Breitbach ZS, Armstrong DW. Enantiomeric impurities in chiral synthons, catalysts, and auxiliaries. Part 3. Tetrahedron Asymmetry. 2006;17(19):2821–32. https://doi.org/10.1016/j.tetasy.2006.10.014.
Manoharan M. 2’-carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim Biophys Acta. 1999;1489(1):117–30. https://doi.org/10.1016/s0167-4781(99)00138-4.
Monia BP, Lesnik EA, Gonzalez C, Lima WF, McGee D, Guinosso CJ, et al. Evaluation of 2′-modified oligonucleotides containing 2’-deoxy gaps as antisense inhibitors of gene expression. J Biol Chem. 1993;268(19):14514–22.
Wave. Patent 10,724,035: Oligonucleotide compositions and methods thereof. 2017.
Prakash TP. An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem Biodivers. 2011;8(9):1616–41. https://doi.org/10.1002/cbdv.201100081.
Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380(24):2307–16. https://doi.org/10.1056/NEJMoa1900907.
Kay C, Collins JA, Caron NS, Agostinho LA, Findlay-Black H, Casal L, et al. A comprehensive haplotype targeting strategy for allele-specific HTT suppression in Huntington disease. Am J Human Genet. 2019. https://doi.org/10.1016/j.ajhg.2019.10.011.
Duyao M, Auerbach A, Ryan A, Persichetti F, Barnes G, McNeil S, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269(5222):407–10. https://doi.org/10.1126/science.7618107.
Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–23. https://doi.org/10.1016/0092-8674(95)90542-1.
Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11(2):155–63. https://doi.org/10.1038/ng1095-155.
Ambrose CM, Duyao MP, Barnes G, Bates GP, Lin CS, Srinidhi J, et al. Structure and expression of the Huntington’s disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet. 1994;20(1):27–38. https://doi.org/10.1007/BF02257483.
Butland SL, Devon RS, Huang Y, Mead C-L, Meynert AM, Neal SJ, et al. CAG-encoded polyglutamine length polymorphism in the human genome. BMC Genom. 2007;8:126. https://doi.org/10.1186/1471-2164-8-126.
Kordasiewicz Holly B, Stanek Lisa M, Wancewicz Edward V, Mazur C, McAlonis Melissa M, Pytel Kimberly A, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–44. https://doi.org/10.1016/j.neuron.2012.05.009.
Warby SC, Montpetit A, Hayden AR, Carroll JB, Butland SL, Visscher H, et al. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J Hum Genet. 2009;84(3):351–66.
Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19(9):774–8.
Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–85. https://doi.org/10.1038/mt.2011.201.
Østergaard ME, Southwell AL, Kordasiewicz H, Watt AT, Skotte NH, Doty CN, et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013;41(21):9634–50. https://doi.org/10.1093/nar/gkt725.
Southwell AL, Skotte NH, Kordasiewicz HB, Østergaard ME, Watt AT, Carroll JB, et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther. 2014;22(12):2093–106. https://doi.org/10.1038/mt.2014.153.
Southwell AL, Kordasiewicz HB, Langbehn D, Skotte NH, Parsons MP, Villanueva EB, et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci Transl Med. 2018. https://doi.org/10.1126/scitranslmed.aar3959.
Kay C, Skotte NH, Southwell AL, Hayden MR. Personalized gene silencing therapeutics for Huntington disease. Clin Genet. 2014;86(1):29–36.
Kay C, Collins JA, Skotte NH, Southwell AL, Warby SC, Caron NS, et al. Huntingtin haplotypes provide prioritized target panels for allele-specific silencing in Huntington disease patients of European Ancestry. Mol Ther. 2015;23(11):1759–71. https://doi.org/10.1038/mt.2015.128.
White JK, Auerbach W, Duyao MP, Vonsattel J-P, Gusella JF, Joyner AL, et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. 1997;17(4):404–10. https://doi.org/10.1038/ng1297-404.
Rodan LH, Cohen J, Fatemi A, Gillis T, Lucente D, Gusella J, et al. A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur J Hum Genet. 2016;24(12):1826–7. https://doi.org/10.1038/ejhg.2016.74.
Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26(3):300–6. https://doi.org/10.1038/81593.
Wang G, Liu X, Gaertig MA, Li S, Li X-J. Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc Natl Acad Sci. 2016;113(12):3359–64. https://doi.org/10.1073/pnas.1524575113.
Dietrich P, Johnson IM, Alli S, Dragatsis I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 2017;13(7): e1006846. https://doi.org/10.1371/journal.pgen.1006846.
Poplawski GHD, Kawaguchi R, Van Niekerk E, Lu P, Mehta N, Canete P, et al. Injured adult neurons regress to an embryonic transcriptional growth state. Nature. 2020;581(7806):77–82. https://doi.org/10.1038/s41586-020-2200-5.
Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, et al. Huntington’s disease alters human neurodevelopment. Science. 2020;369(6505):787–93. https://doi.org/10.1126/science.aax3338.
Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67(3):392–406. https://doi.org/10.1016/j.neuron.2010.06.027.
Agasse F, Mendez-David I, Christaller W, Carpentier R, Braz BY, David DJ, et al. Chronic corticosterone elevation suppresses adult hippocampal neurogenesis by hyperphosphorylating Huntingtin. Cell Rep. 2020;32(1): 107865. https://doi.org/10.1016/j.celrep.2020.107865.
Simpson JM, Gil-Mohapel J, Pouladi MA, Ghilan M, Xie Y, Hayden MR, et al. Altered adult hippocampal neurogenesis in the YAC128 transgenic mouse model of Huntington disease. Neurobiol Dis. 2011;41(2):249–60. https://doi.org/10.1016/j.nbd.2010.09.012.
Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156(5):1072–83. https://doi.org/10.1016/j.cell.2014.01.044.
Parsons MP, Kang R, Buren C, Dau A, Southwell AL, Doty CN, et al. Bidirectional control of postsynaptic density-95 (PSD-95) clustering by Huntingtin. J Biol Chem. 2014;289(6):3518–28. https://doi.org/10.1074/jbc.M113.513945.
Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–38. https://doi.org/10.1016/j.cell.2004.06.018.
Zala D, Hinckelmann M-V, Yu H, da Cunha Marcel ML, Liot G, Cordelières Fabrice P, et al. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152(3):479–91. https://doi.org/10.1016/j.cell.2012.12.029.
Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J Cell Biol. 2006;172(4):605–18. https://doi.org/10.1083/jcb.200509091.
Ochaba J, Lukacsovich T, Csikos G, Zheng S, Margulis J, Salazar L, et al. Potential function for the huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci. 2014;111(47):16889–94. https://doi.org/10.1073/pnas.1420103111.
Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17(3):262–75. https://doi.org/10.1038/ncb3101.
Wong YC, Holzbaur ELF. The regulation of autophagosome dynamics by Huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34(4):1293. https://doi.org/10.1523/JNEUROSCI.1870-13.2014.
Guedes-Dias P, Pinho BR, Soares TR, de Proença J, Duchen MR, Oliveira JMA. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol Dis. 2016;90:51–7. https://doi.org/10.1016/j.nbd.2015.09.008.
Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 2020. https://doi.org/10.1080/15548627.2020.1728096.
Park J, Lee J, Choi C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE. 2011;6(8): e23211. https://doi.org/10.1371/journal.pone.0023211.
Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16(21):2600–15. https://doi.org/10.1093/hmg/ddm217.
Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci USA. 2014;111(8):3032–7. https://doi.org/10.1073/pnas.1314421111.
Ehrnhoefer DE, Southwell AL, Sivasubramanian M, Qiu X, Villanueva EB, Xie Y, et al. HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo. Hum Mol Genet. 2018;27(2):239–53. https://doi.org/10.1093/hmg/ddx394.
Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WMC, Truant R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. 2017;26(2):395–406. https://doi.org/10.1093/hmg/ddw395.
Machiela E, Jeloka R, Caron NS, Mehta S, Schmidt ME, Baddeley HJE, et al. The interaction of aging and cellular stress contributes to pathogenesis in mouse and human Huntington disease neurons. Front Aging Neurosci. 2020. https://doi.org/10.3389/fnagi.2020.524369.
Connolly C, Magnusson-Lind A, Lu G, Wagner PK, Southwell AL, Hayden MR, et al. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience. 2016;325:74–88.
Mattis VB, Tom C, Akimov S, Saeedian J, Østergaard ME, Southwell AL, et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum Mol Genet. 2015;24(11):3257–71. https://doi.org/10.1093/hmg/ddv080.
Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, et al. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96(4):1121–9. https://doi.org/10.1111/j.1471-4159.2005.03605.x.
Munsie L, Caron N, Atwal RS, Marsden I, Wild EJ, Bamburg JR, et al. Mutant huntingtin causes defective actin remodeling during stress: defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. 2011;20(10):1937–51. https://doi.org/10.1093/hmg/ddr075.
Nath S, Munsie LN, Truant R. A huntingtin-mediated fast stress response halting endosomal trafficking is defective in Huntington’s disease. Hum Mol Genet. 2015;24(2):450–62. https://doi.org/10.1093/hmg/ddu460.
McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19(12):2152–62. https://doi.org/10.1038/mt.2011.219.
Grondin R, Kaytor MD, Ai Y, Nelson PT, Thakker DR, Heisel J, et al. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135(4):1197–209. https://doi.org/10.1093/brain/awr333.
Stiles DK, Zhang Z, Ge P, Nelson B, Grondin R, Ai Y, et al. Widespread suppression of huntingtin with convection-enhanced delivery of siRNA. Exp Neurol. 2012;233(1):463–71. https://doi.org/10.1016/j.expneurol.2011.11.020.
Southwell AL, Smith-Dijak A, Kay C, Sepers M, Villanueva EB, Parsons MP, et al. An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum Mol Genet. 2016;25(17):3654–75. https://doi.org/10.1093/hmg/ddw212.
Bečanović K, Nørremølle A, Neal SJ, Kay C, Collins JA, Arenillas D, et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci. 2015;18(6):807–16. https://doi.org/10.1038/nn.4014.
Caron NS, Southwell AL, Brouwers CC, Cengio LD, Xie Y, Black HF, et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2020;48(1):36–54. https://doi.org/10.1093/nar/gkz976.
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. https://doi.org/10.1016/j.addr.2015.01.008.
Schobel SA. Preliminary results from a 15-month open-label extension (OLE) study investigating RG6042 huntingtin protein (HTT) antisense oligonucleotide (ASO) in adults with manifest Huntington's disease (HD). In: CHDI 15th annual HD therapeutics conference. 2020.
Schobel SA. Preliminary results from GENERATION HD1, a phase III trial of tominersen in individuals with manifest HD. In: CHDI 16th annual HD therapeutics conference. 2021.
Roche. Roche provides update on tominersen programme in manifest Huntington's disease. Basel. 2021.
WAVE. Wave life sciences announces topline data and addition of higher dose cohort in ongoing phase of 1b/2a PRECISION-HD2 trial in Huntington’s disease. Cambridge. 2019.
Wave. Wave life sciences provides update on phase 1b/2a PRECISION-HD Trials. Cambridge. 2021.
Wave. Wave life sciences reports fourth quarter and full year 2019 financial results and provides business update. Cambridge. 2020.
Wave. Wave life sciences highlights pipeline progress and expansion leveraging new PN backbone chemistry modifications. Cambridge. 2021.
Viglietta V. A Ph1b/2a study of WVE-003, an investigational allele-selective, mHTT-lowering oligonucleotide for the treatment of early manifest Huntington’s disease, and review of PRECISION-HD results. In: CHDI 16th annual HD therapeutics conference: wave life sciences. 2021.
Wave. Wave life sciences reports third quarter 2020 financial results and provides business update. Cambridge. 2020.
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558–67. https://doi.org/10.1172/jci99081.
Bennett CF, Kordasiewicz HB, Cleveland DW. Antisense drugs make sense for neurological diseases. Annu Rev Pharmacol Toxicol. 2021;61(1):831–52. https://doi.org/10.1146/annurev-pharmtox-010919-023738.
Epping EA, Kim JI, Craufurd D, Brashers-Krug TM, Anderson KE, McCusker E, et al. Longitudinal psychiatric symptoms in prodromal Huntington’s disease: a decade of data. Am J Psychiatry. 2016;173(2):184–92. https://doi.org/10.1176/appi.ajp.2015.14121551.
Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, et al. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79(8):874–80. https://doi.org/10.1136/jnnp.2007.128728.
Scahill RI, Zeun P, Osborne-Crowley K, Johnson EB, Gregory S, Parker C, et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19(6):502–12. https://doi.org/10.1016/S1474-4422(20)30143-5.
Lebel C, Walker L, Leemans A, Phillips L, Beaulieu C. Microstructural maturation of the human brain from childhood to adulthood. Neuroimage. 2008;40(3):1044–55. https://doi.org/10.1016/j.neuroimage.2007.12.053.
Lim B, Grøntvedt GR, Bathala P, Kale SS, Campbell CT, Stengelin M, et al. CSF neurofilament light may predict progression from amnestic mild cognitive impairment to Alzheimer’s disease dementia. Neurobiol Aging. 2021;107:78–85. https://doi.org/10.1016/j.neurobiolaging.2021.07.013.
Halbgebauer S, Steinacker P, Verde F, Weishaupt J, Oeckl P, von Arnim C, et al. Comparison of CSF and serum neurofilament light and heavy chain as differential diagnostic biomarkers for ALS. J Neurol Neurosurg Psychiatry. 2021. https://doi.org/10.1136/jnnp-2021-327129.
Rosenstein I, Axelsson M, Novakova L, Blennow K, Zetterberg H, Lycke J. Exploring CSF neurofilament light as a biomarker for MS in clinical practice; a retrospective registry-based study. Mult Scler (Houndmills, Basingstoke, England). 2021. https://doi.org/10.1177/13524585211039104.
Soylu-Kucharz R, Sandelius Å, Sjögren M, Blennow K, Wild EJ, Zetterberg H, et al. Neurofilament light protein in CSF and blood is associated with neurodegeneration and disease severity in Huntington’s disease R6/2 mice. Sci Rep. 2017;7(1):14114. https://doi.org/10.1038/s41598-017-14179-1.
Szejko N, Picón C, García-Caldentey J, de Yebenes JG, Alvarez-Cermeño JC, Villar LM, et al. Quantification of the light subunit of neurofilament protein in cerebrospinal fluid of Huntington’s disease patients. PLoS Curr. 2018. https://doi.org/10.1371/currents.hd.280c8f9f7d9fa4f7f0c883d9f8e807da.
Southwell AL, Smith SEP, Davis TR, Caron NS, Villanueva EB, Xie Y, et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci Rep. 2015;5(1):12166. https://doi.org/10.1038/srep12166.
Caron NS, Banos R, Yanick C, Aly AE, Byrne LM, Smith ED, et al. Mutant huntingtin is cleared from the brain via active mechanisms in Huntington disease. J Neurosci. 2021;41(4):780. https://doi.org/10.1523/JNEUROSCI.1865-20.2020.
Bacioglu M, Maia Luis F, Preische O, Schelle J, Apel A, Kaeser Stephan A, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron. 2016;91(1):56–66. https://doi.org/10.1016/j.neuron.2016.05.018.
Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27(6):954–63. https://doi.org/10.1038/s41591-021-01382-x.
Neueder A, Landles C, Ghosh R, Howland D, Myers RH, Faull RLM, et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. 2017;7(1):1307. https://doi.org/10.1038/s41598-017-01510-z.
Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA. 2013;110(6):2366–70. https://doi.org/10.1073/pnas.1221891110.
Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125(6):1179–91. https://doi.org/10.1016/j.cell.2006.04.026.
Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci USA. 2005;102(32):11402–7. https://doi.org/10.1073/pnas.0503634102.
Southwell AL, Warby SC, Carroll JB, Doty CN, Skotte NH, Zhang W, et al. A fully humanized transgenic mouse model of Huntington disease. Hum Mol Genet. 2013;22(1):18–34. https://doi.org/10.1093/hmg/dds397.
Acknowledgements
The authors thank Erin Kenzie for figure production and adaptation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Support for manuscript preparation and publication was provided by the University of Central Florida
Conflict of interest
A.L.S has performed preclinical sponsored research in collaboration with IONIS Pharmaceuticals and Wave Life Sciences at her current and previous academic institutions. This work has been published or publicly presented and did not include investigation of the ASO molecules discussed here. M.E.R declares no conflicts of interest. The authors declare no competing interests.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
M.E.R wrote the manuscript, M.E.R. and A.L.S. edited and revised the manuscript.
Rights and permissions
About this article

Cite this article
Rook, M.E., Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs 36, 105–119 (2022). https://doi.org/10.1007/s40259-022-00519-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-022-00519-9