
Abstract
Biosimilars are highly similar versions of approved branded biologics. Unlike generics, they are not exact replicas of reference products. Minor differences between biosimilars and reference products in some aspects are expected; likewise, biosimilar products will differ from each other. The objective of this review is to discuss the challenges associated with the development and approval of biosimilar products that are unique because of their complex structure and specialized manufacturing processes, which can impact not only efficacy but also immunogenicity and safety. Regulatory guidelines recommend a totality-of-evidence approach focused on stepwise development that involves demonstration of structural similarity and functional equivalence. Structural and functional characteristics of the proposed biosimilar are compared with the reference product; similarity of these functions forms the foundation of the biosimilar development program, including potential animal studies, a human pharmacokinetics/pharmacodynamics equivalence study, and a clinical study to confirm similar efficacy, safety, and immunogenicity. The clinical study should be performed in a sensitive population using appropriate endpoints to allow detection of any clinically meaningful differences between the biosimilar and the reference product if such differences exist. In conclusion, development of biosimilars is focused on the minimization of potential differences between the proposed biosimilar and reference product and the establishment of a robust manufacturing process to consistently produce a high-quality biosimilar product.
Similar content being viewed by others

Development of biosimilars presents considerable challenges due to their complex structure and specialized manufacturing processes that could have clinical implications; similarity of structural and functional characteristics of the proposed biosimilar to the reference product forms the foundational first step in the totality of evidence for biosimilarity demonstration. |
The goal of the biosimilar clinical development program is not to demonstrate efficacy and safety per se but rather to confirm similarity with the reference product based on pharmacokinetic/pharmacodynamic equivalence and a confirmatory comparative pivotal clinical study in a representative indication evaluating safety, efficacy, and immunogenicity. |
Regulatory guidance allows for extrapolation to all indications of use for which the reference product is approved with scientific justification centered around the totality of evidence that supports similarity between the proposed biosimilar and the reference product based on same mechanisms of action while simultaneously considering the physiology of each disease. |
1 Introduction
Biologics, biological medicines derived from genetically modified living organisms, represent a large proportion of approved therapies for cancer and chronic inflammatory diseases. The development of recombinant protein and antibody therapies have led to the introduction of additional options to address previously unmet therapeutic needs. With the expiration of patents on several originator biologics, the EU pioneered the establishment of the regulatory framework for the development and approval of biosimilars with their first biosimilar approval in 2006 for human growth hormone. Since then, several biosimilars have entered the European market, including several somatropins, epoetins, and more recently, monoclonal antibodies (mAbs) (Table 1). To improve access to biologics, the US Congress passed the Biologics Price Competition and Innovation Act of 2009, which authorized the US Food and Drug Administration (FDA) to oversee an abbreviated and expedited pathway [351(k) pathway] for the approval of biosimilars [1]. The FDA guidelines for biosimilar development are quite similar to those of the European Medicines Agency (EMA). The FDA approved its first biosimilar, Zarxio™ (filgrastim-sndz), in March 2015 and has since approved two mAb biosimilars, Inflectra® (infliximab-dyyb) and AMJEVITA™ (adalimumab-atto), as well as Erelzi™ (etanercept-szzs).
The EMA defines a biosimilar as a biological medicine that is similar to another biological medicine that has already been authorized for use in terms of quality characteristics, biological activity, safety, and efficacy, based on a comprehensive comparability exercise [2, 3]. According to the FDA definition [4,5,6,7], a product is a biosimilar if data from analytical, animal, and clinical studies show the product to be highly similar to the reference product, notwithstanding minor differences in clinically inactive components, and if there are no clinically meaningful differences in terms of safety, purity, and potency.
Although biosimilars are similar to originator reference molecules, they are not analogous to generic drugs because they are not identical to the originator biological agents. Small-molecule generic drugs have relatively simple and well defined chemical structures, whereas biologics, such as recombinant proteins and mAbs, are large complex molecules. In contrast to small-molecule drugs, which are manufactured by chemical synthesis, the manufacturing processes for biologics involve living systems, such as microbial and animal cells, typically cultivated to adapt to unique growth environments. The living systems are sensitive to manufacturing processes; therefore, each biosimilar is expected to differ from the originator as well as from other biosimilars [8, 9].
There have been several recent reviews on biosimilars [10,11,12,13,14]; the purpose of this review is to discuss the science of biosimilar development and approval and highlight challenges. Although there are several pharmacoeconomic considerations associated with the use of biosimilars, these will not be addressed here.
2 Development of Biosimilars
The challenge of producing a biosimilar is quite different from that of reproducing a small molecule to develop a generic drug product. The design and development of a successful biosimilar requires an in-depth understanding of the structure and function of the reference (originator) product to establish a target quality profile that can be used to evaluate any potential analytical differences and their relationship to function. It is also uniquely important to define critical quality attributes (CQAs), attributes that impact pharmacokinetics (PK), safety, or efficacy, for a proposed biosimilar via thorough analytical characterization. CQAs are defined as physical or biological properties of a product that should be characterized and controlled within an appropriate range to ensure product quality. Although proposed biosimilars are expected to have the same amino acid sequence as the reference molecule, low level sequence variants may be detected by highly sensitive methods. These variants may be the result of mutations in the DNA or misincorporation due to mistranslation or improper tRNA acylation [15]. Additionally, biological products are subject to cell line-dependent post-translational modifications (PTMs) during cellular expression, including modifications at the N- or C-terminus such as amino acid cleavage, methylation, N-acetylation, and, most important to biological function, glycosylation. Purity and final product profiles are also influenced by purification methods, formulation and storage conditions, and container-closure systems. Manufacturers of originator products use proprietary growth and purification conditions and specially adapted cell lines for their processes; therefore, knowledge of the protein sequence or cells used by the originator is not sufficient for a biosimilar sponsor to produce the same biologic product. Differences in structure between proposed biosimilar and reference product need to be minimized because even subtle differences can potentially affect PK, efficacy, safety, and immunogenicity [16,17,18].
The stepwise demonstration of biosimilarity includes in vitro analytical testing, nonclinical comparative pharmacology, toxicology, PK testing, and one or more clinical trials to confirm quality, efficacy, and safety of the proposed biosimilar as compared with the reference product (Fig. 1). The objective of analytical testing is to establish similarity assessment criteria based on the reference product profile, followed by a comparative assessment of the biosimilar candidate and the reference product. The knowledge gained from understanding the originator product is used to develop a proposed biosimilar product that has similar structural and functional characteristics as the reference product. The next critical component of the biosimilarity exercise, assuming the molecule demonstrates high structural and functional similarity, is a targeted clinical development program in which the PK, efficacy, safety, and immunogenicity of the proposed biosimilar are compared with that of the reference product and demonstrated to be similar. Regulatory guidelines recommend analytical studies to serve as the foundation for establishing similarity to the reference product. A high degree of analytical similarity, with special emphasis on all biological functions being similar, provides justification for the reduced regulatory requirements with respect to preclinical and clinical studies, which further facilitates the overall abbreviated approval process for biosimilars. These requirements suggest that the development of a high-quality biosimilar necessitates significant technical capability and manufacturing expertise.

Stepwise process for biosimilarity demonstration. PD pharmacodynamics, PK pharmacokinetics
2.1 Production of the Proposed Biosimilar Molecule
Biosimilars must be similar to the reference product in structure and function. Process optimization toward similarity and precise control during manufacturing to maintain similarity is important for the quality of biosimilars. Development of a biosimilar begins with transfection of a cell line, typically one that is different from that used by the originator, with a DNA vector encoding the product; however, starting with the correct amino acid sequence does not guarantee that the biosimilar product will have biological functions similar to the reference product. The use of quality-by-design strategies allows a proposed biosimilar to achieve high similarity of the complex features, ensuring quality and safety. The product and process knowledge includes an understanding of the effect of normal operating process parameters, variability due to source raw materials, as well as the equipment and manufacturing facility, on product quality. Sponsors of biosimilar products should consider all relevant characteristics of the proposed molecule, such as the primary, secondary, tertiary, and higher-order structure, PTMs, and biological activity. Risk assessment tools enable linking and ranking of quality attributes to product safety and efficacy, which is augmented by historical and cumulative knowledge about the desired quality attributes for the biosimilar candidate gained from an understanding of the reference product. For example, to develop a biosimilar to a mAb that has antibody-dependent cellular cytotoxicity (ADCC) activity, knowledge of the relevant PTMs is used to appropriately select the host cell type for the final clone and optimization of the manufacturing process. This task is complicated by the added challenge of having to evaluate multiple CQAs important for safety and efficacy (i.e., ensuring all functions are similar), which may be differentially affected by attributes such as the glycan profile. Based on this information, media and production format are selected and cell culture and purification processes are refined such that multiple CQAs are ensured to be similar to the reference product. Formulation is then developed to ensure appropriate stability and robustness of the product.
Born and Fung [19] presented two case studies highlighting the challenges and importance of host cell line selection to ensure similar CQAs between the proposed biosimilar and the reference product. In the first case, the production of a proposed biosimilar mAb in a Chinese hamster ovary (CHO) expression system, similar to the reference product, was examined. In the second, production of a proposed biosimilar in a different host expression system from that of the reference product was evaluated; a mAb produced in a murine myeloma cell line was compared with a mAb produced in CHO cells. The glycan profile of reference product and proposed biosimilar mAb were assessed, as were the resulting ADCC and complement-dependent cytotoxicity activity of the molecules. Characterization of mAb1 demonstrated that two mAbs of the same amino acid sequence and expressed in the same parental expression system could result in significant differences in glycan distribution. These differences in glycans could not be resolved with process development and had significant impact on the functional activity of mAb1 (Fig. 2a). In contrast, characterization of mAb2 illustrated the ability to ensure similar functional activity between an originator mAb and proposed biosimilar, even with differences in choice of cellular expression system and the expected underlying differences in glycan distribution (Fig. 2b). These cases elucidated that use of the same parental cell line as the reference product does not guarantee that CQAs will be similar in a proposed biosimilar and that different host cell systems can potentially still have similar functional attributes.

a ADCC activity differs for mAb1 despite being produced in the same cell type. Three different batches of proposed biosimilar mAb1 were produced by process 1 (red and green) or process 2 (orange) and tested for ADCC activity. Neither process condition was able to produce an antibody with ADCC activity similar to that of the originator mAb1 (blue). In contrast, b demonstrates similar ADCC activity for mAb2 despite being expressed in differing cell systems. The graph compares US-sourced (black) reference product and EU-sourced (blue) reference product produced in murine cells, compared with the mAb2 proposed biosimilar (red) produced in CHO cells. Ab antibody, ADCC antibody-dependent cellular toxicity, CHO Chinese hamster ovary, mAb monoclonal antibody
Most therapeutic proteins are glycosylated, and glycosylation can influence the biological activity of a protein by affecting binding to complement and Fcγ receptors, ultimately impacting effector functions [20,21,22]. Glycan structure–activity relationships are complex and need to be well characterized to deliver a product with similar functional activity. For example, it is critical to ensure that the glycan attributes are characterized in a sensitive manner and that any observed differences do not result in differences in FcγRIIIa binding, particularly with regard to ADCC activity. In the mAb2 case presented by Born and Fung, although the total afucosylation was different between the two mAbs, because the level of hybrid and complex glycans were different in the two cell lines, the effector function was similar [19]. High mannose and β-galactosylation were similar between the two mAbs following extensive process optimization. Clone selection and process optimization allowed for similarity in effector function between two mAbs produced in different cell expression systems despite the expected differences in glycan profiles.
Sustainability and increasing confidence in biosimilar products for human therapeutics relies on robust standards for quality, safety, and efficacy. Meeting these standards begins with a demonstration of analytical and functional similarity between products, which is based on the foundation of understanding the mechanism of action and structure–function relationships. Furthermore, the proposed biosimilar should be compared with the reference product using analytical methods demonstrated to be sufficiently sensitive to detect differences. Although some physiochemical differences are to be expected, bioanalytical differences should not exist. If bioanalytical differences are identified, additional studies should be conducted to determine if the observed difference is clinically meaningful. Advances in cell culture engineering, state-of-the-art bioprocessing, and high-resolution analytics have contributed to the ability to develop and manufacture molecules that can be determined to be highly similar to the originator biologic products with a high degree of confidence.
2.2 Demonstration of Analytical Similarity
A meaningful assessment of analytical similarity requires extensive and robust comparative studies using state-of-the-art analytical techniques. The capability of analytical methods concerning their resolution and reliability, which directly influences the quality of results demonstrating high similarity of the biosimilar candidate to the reference product, also needs to be established. Frequently, orthogonal techniques are used to elucidate and verify any structural and functional differences between the biosimilar molecule and the reference product.
A comprehensive and well-designed analytical similarity assessment demonstrating that comparative results lie within the prespecified assessment criteria, established based on reference product profiles, can significantly reduce the residual uncertainty of biosimilarity. Physicochemical and biological properties should be demonstrated to be highly similar between the proposed biosimilar and reference product. This assessment typically comprises a series of comparative studies and examines product quality attributes in multiple analytical disciplines, including primary structure, higher order structure, biological properties, product-related substance and impurities, process-related impurities, particles and aggregates, general properties, and thermal stability profiles. Assessment of the primary structure identifies attributes related to the amino acid sequence and all PTMs, including glycans. Examinations of the higher order structure evaluate the integrity of the secondary, tertiary, and quaternary structure, whereas examinations of the biological properties include review of target and Fc receptor binding (as relevant) as well as functional assays that reflect the mechanisms of action of the molecule.
Characterization of product and process-related impurities that result from the different manufacturing processes used to produce the proposed biosimilar aids in ensuring product safety. Additionally, the identity and quantity of product-related variants may change over the course of the product shelf life. For this reason, product-related variants need to be evaluated with the same consideration as stability-indicating properties (Fig. 3). Assessment of particles and aggregates identifies any subvisible and submicron particles and characterization of these aggregates of various sizes identifies any impurity that could have immunogenicity and safety concerns. General property assessments measure properties of the finished drug product, such as strength and formulation. Evaluation of thermal stability examines the forced degradation profiles and degradation products. Figure 4 depicts the various physicochemical and biological properties that should be considered for detailed characterization of any biologic. Examination of multiple batches of originator product and the proposed biosimilar is necessary to understand the process variability of the two products. The total number of batches included in the similarity assessment should provide sufficient power for statistical analysis of results to meet regulatory requirements [2].

Characterization of product-related substances and impurities using stability-indicating assay. SE-HPLC size exclusion high performance liquid chromatography

Schematic representation of exemplary quality attributes of a biosimilar monoclonal antibody. C-Term Lysine C-terminal lysine, Fab fragment antigen-binding, Fc fragment crystallizable, FcRn neonatal Fc receptor, FcγR Fc-gamma receptor, N-Term Lysine N-terminal lysine, N-Term Pyro Glu N-terminal pyroglutamate
Analytical similarity assessment is a repetitive and iterative process conducted throughout biosimilar product development, with the goal to increase knowledge and confidence of analytical similarity of the proposed biosimilar with the reference molecule (Fig. 5) [2, 5, 6, 8, 16]. This includes, in particular, a determination of the biological activity of the proposed biosimilar with respect to the currently understood mechanism(s) of action of the reference molecule. If the molecule contains multiple functional domains (e.g., mAb), the binding affinity and specificity at individual domains and the combined biological functions should be compared with those of the reference molecule. For example, target binding and potency mediated by the F(ab) domain should be similar, as should binding to Fc receptors and effector functions mediated by the F(c) domain.

Iterative steps in the process development and analytical similarity assessment for a proposed biosimilar product. DP drug product, DS drug substance
An important consideration during the development of biosimilars is the differentiation between comparability and biosimilarity. Comparability refers to the comparative assessment of characteristics of the biologic product after a specific change in the manufacturing process and is implemented by a manufacturer for their product. The implementation of such a change is supported by comprehensive knowledge and development history of the product. Biosimilarity is a more recent concept whereby the sponsor develops a molecule similar in structure and function to the reference molecule, using a different cell line and process without knowledge of the development history of the reference product; demonstration of biosimilarity is based on a comparative characterization of this product, produced by a different manufacturer, with the reference product [23].
2.3 Preclinical Considerations
In vivo disease models can be informative in characterizing comparative dose-response efficacy if the products cross-react with the target in a relevant species. The dose–response relationship can be particularly informative for assessing equivalence in activity when doses on the steep part of the dose–response curve can be tested. In particular, this may be important for the development of oncology products for which there are no pharmacodynamics (PD) markers, and may increase confidence in similar antitumor activity, thereby increasing the totality of evidence (TOE) and reducing the residual uncertainty of clinical benefit. In such situations, animal models with human tumors can be used to evaluate multiple dose levels of the proposed biosimilar in direct comparison with the reference to demonstrate equivalent antitumor activity. Studies of the proposed biosimilar in preclinical disease models may also support similarity between the products in extrapolated indications.
Preclinical studies for comparison of in vivo pharmacology (PK and PD), toxicology, and immune response should be considered, specific to the biosimilar product in development and its cross-reactivity in other species. Toxicology studies can be conducted if a pharmacologically relevant species is available, but are not particularly relevant in the absence of cross-reactivity. Preclinical toxicology studies in appropriate test species can remove some uncertainty before human testing and can include PK/PD assessments. With sufficient confidence in analytical and in vitro pharmacologic similarity, the use of animals could be minimized or eliminated. For instance, an important step in evidence generation may be a repeat-dose toxicology study without the need for recovery animals, or only testing a high dose level previously tested by the originator to determine similar known toxicities and any potential unexpected toxicity. Ideally, this can be done in a single sex to minimize the use of animals if there are no questions of sex-specific toxicity [2]. In vitro assessments such as cell-based bioactivity assays may be used rather than in vivo studies. For example, an in vitro bioassay is a sensitive method for assessing the potential for inducing cytokine release syndrome in vivo [24]. If sufficiently sensitive and specific, such assays can affect the amount and type of additional animal or clinical data that may be needed to establish biosimilarity. A comparison of preclinical PK and PD may be useful in reducing residual uncertainty regarding similarity, and such a study may be conducted as a comparative single-dose study or incorporated into a single preclinical toxicity study if appropriate. Immunogenicity may also be evaluated in animal studies; results may aid in detection of differences between the proposed biosimilar and reference product as opposed to predicting clinical similarity in immunogenicity.
2.4 Clinical Considerations
The goal of the clinical development program for a biosimilar is to demonstrate the absence of any clinically meaningful difference relative to the reference molecule. The extent of the clinical program depends on the degree of similarity demonstrated in preclinical testing, including structural, functional, and animal studies. According to the FDA guidelines [6], if there are no residual uncertainties with respect to clinically meaningful differences between the proposed biosimilar and the reference product, clinical efficacy studies may not be necessary. However, it is believed that studies of human safety and immunogenicity would still need to supplement the overall evidence since these parameters cannot be predicted outside of a clinical study in an informative population with appropriate duration of exposure and follow-up as well as the use of sensitive assays. Moreover, for many mAbs, clinical trials would likely be obligatory because PD efficacy markers often do not exist. Comparative clinical efficacy and safety studies will also likely be mandatory for other large, structurally complex heterogeneous biologics (such as fusion proteins and mAbs) to confirm comparable efficacy and minimize the risk of adverse outcomes [2, 6, 25].
2.4.1 Human Pharmacology (Pharmacokinetics and Pharmacodynamics)
Clinical development of a proposed biosimilar starts with a study designed to demonstrate PK similarity of the proposed biosimilar and the reference product; human PK and/or PD studies are fundamental components in supporting biosimilarity [2, 6, 26]. Selection of an appropriate study population (patients versus healthy subjects) for these studies should be scientifically justified [6]. Whenever possible, it may be best to conduct the study in healthy subjects to ensure a homogenous population comprising immune-competent subjects who are not receiving any concomitant medications, thereby allowing a sensitive comparison of the proposed biosimilar and reference product. When it is not feasible to conduct the study in healthy subjects, the study may be conducted in a representative patient population. A general standard to establish PK bioequivalence is for the 90% confidence interval of the geometric mean ratio to be within 80–125% for overall exposure (e.g., maximum serum concentration and area under the serum concentration-time curve) [27], to demonstrate that the proposed biosimilar product has equivalent exposure to the reference product. This is critical to the abbreviated development program for biosimilars because, when combined with demonstration of highly similar analytical and functional activity, it allows the pivotal clinical studies to be conducted directly using the same therapeutic dose as the reference, thus rendering phase II studies unnecessary. Human PD studies should be conducted if a marker that is relevant to the mechanism of action is available and provides information regarding clinical efficacy.
2.4.2 Efficacy and Safety
Efficacy and safety studies should be performed in populations that are sensitive enough to detect clinically meaningful differences between the proposed biosimilar and the reference product if such differences exist [6]. The goal of the comparative clinical studies is to demonstrate that the biosimilar candidate has neither decreased nor increased efficacy, and does not have an increased safety risk compared with the reference product. The most straightforward design is one in which the null hypothesis, based on a prespecified equivalence margin, is a two-sided test procedure that demonstrates that the proposed biosimilar is neither inferior nor superior to the reference product [28]. The margins should be scientifically justified and adequate to enable detection of clinically meaningful differences in effectiveness, if a difference exists. An acceptable equivalence margin is chosen based on historical data and relevant clinical and statistical considerations for each given molecule. The historical data provide an estimate of the effect size and the relevant clinical consideration either confirms that the statistically derived margin is clinically meaningful or needs further calibration. The efficacy endpoint can be that of clinical benefit, or alternatively, a meaningful surrogate for efficacy. Ideally, safety is assessed in the same study as efficacy; the choice of patient population should also consider sensitivity for detection of differences with respect to safety. Generally, this may be a population for which the investigational product is used as monotherapy.
An additional consideration is the use of surrogate endpoints that can also play a key role in biosimilar development. For example, whereas overall survival (OS) is considered a gold standard for proving clinical benefit in oncology, it is often not a practical endpoint because it is not necessary for biosimilars to reestablish clinical benefit per se; instead, the endpoint needs to be sensitive enough to detect a difference in activity if one exists. Overall response rate (ORR) and complete response could be suitable endpoints [29].
Another example of an appropriate surrogate endpoint is pathologic complete response (pCR) in neoadjuvant breast cancer (BC) [29]. A pCR is usually defined as the absence of residual invasive disease in the breast and axillary lymph nodes at the completion of neoadjuvant treatment. Based on results from several individual studies and further confirmation from a recent large meta-analysis, it is now established that patients who achieve pCR with neoadjuvant therapy tend to have a better prognosis than patients who had residual invasive disease at the end of treatment [30]. The meta-analysis also showed that in patients with more aggressive cancers, such as triple-negative breast cancer (TNBC) and especially HER2-positive BC, the benefit of achieving pCR in terms of recurrence and survival is much greater. Patients achieving pCR had half the risk of distant disease recurrence compared with patients who did not achieve pCR, and a 36% improvement in OS. Furthermore, patients with more aggressive cancer subtypes, such as hormone receptor-negative, HER2-positive, and TNBC, experienced a 75% improvement. Jackisch et al. performed a meta-analysis of trastuzumab clinical trial data in an effort to develop an optimal approach for the demonstration of similarity between a proposed biosimilar and reference product [31]. Results of this analysis suggested that total pCR in HER2-positive BC is a more sensitive endpoint than ORR in HER2-positive metastatic BC.
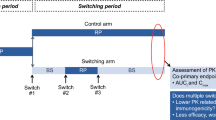
Clinical study designs for evaluation of biosimilars may include a single switch or transition phase in which the study population in the comparator arm is re-randomized to either receive the proposed biosimilar or continue in the comparator arm. The key objective is to ensure that there are no immunogenicity concerns after switching from the reference product to the proposed biosimilar.
2.4.3 Immunogenicity
The FDA explicitly stated that “immunogenicity remains a critical factor when assessing biosimilarity, and the FDA will evaluate immunogenicity in a risk-based manner” [1]. Most biopharmaceuticals can induce immune responses, which in many cases do not have clinically relevant consequences. The immune response can include the development of antidrug antibodies that may bind to the drug with no consequence, increase clearance, or reduce its effectiveness, and develop neutralizing antibodies that eliminate activity. The most severe circumstance is the cross-reaction of antidrug antibodies with an endogenous protein, eliminating its critical function and potentially causing harm.
The extent of immunogenicity can vary due to changes in the manufacturing processes or interactions with packaging components; for example, in the cases of pure red cell aplasia (PRCA) in patients receiving a brand of epoetin (or recombinant human erythropoietin) approved in Europe [32] that were attributed to formulation change. Additionally, a biosimilar being tested for subcutaneous use was found to have a significantly increased rate of PRCA, leading to early study termination [33]. Immune responses may affect both safety and effectiveness of the product by altering PK, inducing anaphylaxis, or promoting the development of neutralizing antibodies. Thus, establishing an immune response that is similar to that seen with the reference product is a key element in the demonstration of biosimilarity. The clinical trial should be of appropriate duration to allow for development of a detectable immune response. Additionally, adequate pharmacovigilance programs are necessary to ensure that adverse events are appropriately attributed.
2.4.4 Extrapolation
The concept of extrapolation of indications allows for a biosimilar product to potentially be labeled for use in indications approved for the reference product that were not studied as part of the clinical development program of the biosimilar. This is an important consideration because if clinical trials were to be conducted in each indication, the breadth of biosimilar development programs would effectively negate the advantages of an abbreviated approval pathway based on developing a product that is highly similar to a reference product with an established risk–benefit profile [28]. Regulatory guidelines state that extrapolation is allowable provided scientific justification is available [2, 6]. Scientific justification should address the mechanism of action, biodistribution, immunogenicity, and expected toxicities in each patient population; these factors should be well understood based on reference product knowledge in each indication. Additionally, any other factor that may affect the safety, effectiveness, or immunogenicity of the product in each condition of use should be appropriately addressed for each indication in the justification for extrapolation. The justification for extrapolation of claims of safety and efficacy in all indications is supported by the TOE demonstrating biosimilarity of the biosimilar candidate and the reference product. Although there is considerable caution with respect to this concept, Weise et al. have noted that extrapolation is a logical consequence of the biosimilar concept that has been successfully implemented [34].
3 Regulatory Considerations
The implementation of an abbreviated licensure pathway for biologic products can pose various challenges given the scientific and technical challenges that may be associated with larger and more complex structures of biologic products. The EMA has pioneered the issuance of guidelines for the development and approval of biosimilars since 2004 [35]; the FDA guidelines are based on similar principles.
Both agencies acknowledge the complexity of biosimilar development, noting that all biosimilar candidates will have unique aspects specific to the molecule and will require targeted case-by-case development programs. Additionally, the World Health Organization has also developed guidelines to support the worldwide development of biosimilars; these guidelines follow the same principles as the EMA and FDA guidelines [8, 36]. As the health authorities strive to align the regulatory requirements, it is important to consider a number of important regulatory concepts supporting the biosimilar development program.
The US and EU laws define the reference product as that approved in the local jurisdiction. Thus, for approval in the US, the proposed biosimilar must be shown to be similar to the reference product approved in the US; for approval in the EU, it must be shown to be similar to the reference product approved in the European Economic Area [3, 4]. However, acknowledging the complexity and expenses associated with biosimilar development, both agencies have taken steps to facilitate global development programs and implemented provisions to allow the use of foreign-sourced comparators in comparative clinical studies, provided a scientific rationale exists to bridge the foreign product to the one approved in the local jurisdiction. From a regulatory and legislative perspective, acceptability of reliance on the clinical data generated utilizing a foreign-sourced comparator is contingent upon successful establishment of the scientific bridge [3, 4]. The “scientific bridge” between the local and the foreign-sourced originator product should consist of comprehensive analytical similarity assessment of the biosimilar candidate versus both comparators. Additionally, a three-arm PK similarity study in which bioequivalence is established between the biosimilar candidate and each respective comparator as well as between the two comparator arms should complete the bridge.
An additional regulatory consideration for biosimilars that is unique to the US is the determination of interchangeability. According to the law, an interchangeable biosimilar product is one in which the product has been shown first to be biosimilar to the reference and is expected to produce the same clinical result as the reference product in any given patient. For a biological product that is administered to an individual more than once, the risk in terms of safety or diminished efficacy due to alternating or switching between the biosimilar and reference products should not be greater than the risk of using the reference product without such alternation or switch [37].
The FDA has issued draft guidance for demonstration of interchangeability for biosimilars [38]; the guidance also adopts the TOE and stepwise approach. This guidance would require at least one study involving three or more switches between the biosimilar and its US-licensed reference product demonstrating that the biosimilar produces the same clinical result for all reference product indications of use. The sponsor may provide a scientific rationale to extrapolate data supporting interchangeability in one of the remaining conditions of use for which the reference product is licensed. It is recommended that the study should be conducted in appropriate patient populations in one or more indications of use with endpoints that assess the impact of switching on PK (and PD if a suitable marker is available). This is because PK (and PD) are expected to be more sensitive to potential changes in immunogenicity due to any potential residual differences that can stimulate an immunogenic response when switching between the biosimilar and reference product. Of note, the guidance states that postmarketing data from products first licensed and marketed as biosimilars that lack corresponding data derived from a prospective switching study or studies would not be sufficient to support a demonstration of interchangeability. The guidance further emphasizes the importance of adequate pharmacovigilance mechanisms for postmarketing safety monitoring of interchangeable products.
The EU guidance on biosimilars does not include specific requirements for interchangeability. Although most biosimilars are approved centrally by the EMA, individual member states make substitution policies.
Another consideration for the successful adoption of biosimilars includes naming criteria and safety monitoring or pharmacovigilance. The FDA has issued a guidance that discusses the use of distinguishable names by adding a unique 4-letter suffix to the international nonproprietary name (INN) for each biosimilar product to avoid inadvertent substitution by a pharmacist based on lack of specificity of the product name, particularly when products have not been approved as interchangeable [39]. Moreover, distinguishable names are important from a pharmacovigilance standpoint to ensure the appropriate attribution of adverse events to the correct manufacturer [39].
4 Key Challenges
The key tenet in biosimilar development is the practical implementation of the concept of TOE. What does this mean? What is the stepwise process? While determination of structural similarity and functional equivalence of the proposed biosimilar to the reference product is the obligatory first step, the details of what is entailed in this exercise are dependent on the molecule. The number of tests performed, as well as the sensitivity of those tests to detect potential differences to conclude that a thorough and complete characterization has been performed with no residual uncertainty, is determined by each sponsor based on the specific molecule. The identification of CQAs of a molecule and the establishment of a product quality profile are also at the discretion of the sponsor. For example, as discussed here, the use of the same expression system does not guarantee structural and functional similarity; likewise, the use of different expression systems does not imply a lack of biosimilarity in structure and function of the proposed biosimilar. Design and interpretation of clinical studies, statistical analysis plans, sample size calculations, selection of appropriate endpoints, and a sensitive population such that similarity between the proposed biosimilar and the reference product is demonstrated in a way that there are no clinically meaningful differences, are all points that deserve due consideration; the definition of suitable endpoints and sensitive population is subject to interpretation. Defining statistical margins and confidence limits for equivalence testing is another vital point. It is important to consult regulatory agencies at early stages of the development program to reach agreement on the development strategy, including choice of assays for analytical similarity assessments, clinical study designs, study endpoints, choice of population, and statistical approach to establish the TOE. It may also be important to engage with the agencies as critical data elements become available to minimize residual uncertainty with respect to the establishment of biosimilarity. Additionally, based on the totality of data provided, extrapolation to all or some indications may or may not be appropriate. Differences in data packages pose a considerable challenge and must be appropriately understood and evaluated by stakeholders.
Another consideration in understanding the stepwise process is the implementation of the tailored approach to the development of a biosimilar product. While the actual execution of each step in the development program may follow the stepwise process, the design of each of these steps remains independent of each other. For example, the study designs for the PK and clinical studies are mutually exclusive. The design of the clinical study with respect to the choice of the patient population, endpoints, and statistical margins is typically considered independently from the results of the previous steps of development, such as analytical and PK similarity. All these factors pose unique challenges, and although the agencies have outlined a theoretical stepwise approach to the development of biosimilars, the practical implementation and the specifics of such an approach remain dependent on decisions by each manufacturer or sponsor.
5 Conclusions
Biosimilars are different from generics in that the product attributes of a proposed biosimilar are not identical to those of the reference product. Instead, the product attributes of the biosimilar are expected to be highly similar to those of the reference product, with only minor differences that do not affect clinical activity. Therefore, the development and regulatory considerations are appropriately different from those for generic drugs (Table 2). The development and regulation of biologics present considerable challenges due not only to their complex nature and production process but also to specific safety concerns linked to immunogenicity potential and immunological activity of complex biologics. Considerable experience and expertise are required for the development of a robust biosimilar that can be reproduced with predefined and established quality characteristics to ensure that patients receive high-quality treatments.
An important consideration in the adoption and use of biosimilars not discussed here is related to pharmacoeconomics; there have been a few recent publications on this topic [40,41,42,43,44]. As more biosimilars appear on the market and more experience is gained, pharmacoeconomic evaluations would become center stage and provide additional insights into the place of biosimilars in the overall treatment paradigm.
Biosimilars as therapeutic alternatives are an independent category of products and should be treated differently from generics. The complexities associated with their development and approval for use and the specifics of each product warrant a targeted approach and require expertise and rigorous standards to maintain high-quality options for patients.
References
Kozlowski S, Woodcock J, Midthun K, Sherman RB. Developing the nation’s biosimilars program. N Engl J Med. 2011;365(5):385–8.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. London, UK. 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf. Accessed 30 March 2016.
European Medicines Agency. Guideline on similar biological medicinal products. London, UK. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Accessed 30 March 2016.
US Food and Drug Administration. Guidance for industry: biosimilars: questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009. Rockville, MD. 2015. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM444661.pdf. Accessed 30 March 2016.
US Food and Drug Administration. Guidance for industry: quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. Rockville, MD. 2015. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf. Accessed 30 March 2016.
US Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. Rockville, MD. 2015. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 30 March 2016.
US Food and Drug Administration. Guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Rockville MD. 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed 29 April 2016.
Chow SC, Wang J, Endrenyi L, Lachenbruch PA. Scientific considerations for assessing biosimilar products. Stat Med. 2013;32(3):370–81.
Abraham I, Sun D, Bagalagel A, Altyar A, Mohammed A, Tharmarajah S, et al. Biosimilars in 3D: definition, development and differentiation. Bioengineered. 2013;4(4):203–6.
Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin JH, Weise M, et al. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012;30(12):1179–85.
Tsiftsoglou AS, Ruiz S, Schneider CK. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27(3):203–11.
Macdonald JC, Hartman H, Jacobs IA. Regulatory considerations in oncologic biosimilar drug development. mAbs. 2015;7(4):653–61.
Dorner T, Strand V, Cornes P, Goncalves J, Gulacsi L, Kay J, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974–82.
Lemery SJ, Esteva FJ, Weise M. Biosimilars: here and now. Am Soc Clin Oncol Educ Book. 2016;35:e151–7. doi:10.14694/EDBK_155954.
Yang Y, Strahan A, Li C, Shen A, Liu H, Ouyang J, et al. Detecting low level sequence variants in recombinant monoclonal antibodies. mAbs. 2010;2(3):285–98.
Barnes HJ, Ragnarrson G, Alvan G. Quality and safety considerations for recombinant biological medicines: a regulatory perspective. Int J Risk Saf Med. 2009;21:13–22.
Kuhlmann M, Covic A. The protein science of biosimilars. Nephrol Dial Transpl. 2006;21(Suppl 5):v4–8.
Colbert RA, Cronstein BN. Biosimilars: the debate continues. Arthritis Rheum. 2011;63(10):2848–50.
Born T, Fung V, editors. Analytical and functional assessments when developing biosimilar candidates. Paris: European League Against Rheumatism (EULAR); 2014.
Kuhlmann M, Covic A. The protein science of biosimilars. Nephrol Dial Transpl. 2006;21(suppl 5):v4–8.
Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. mAbs. 2013;5(5):621–3.
Flynn GC, Chen X, Liu YD, Shah B, Zhang Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol Immunol. 2010;47(11–12):2074–82.
Declerck P, Farouk-Rezk M, Rudd PM. Biosimilarity versus manufacturing change: two distinct concepts. Pharm Res. 2016;33(2):261–8.
Walker MR, Makropoulos DA, Achuthanandam R, Van Arsdell S, Bugelski PJ. Development of a human whole blood assay for prediction of cytokine release similar to anti-CD28 superagonists using multiplex cytokine and hierarchical cluster analysis. Int Immunopharmacol. 2011;11(11):1697–705.
Nick C. How can the biosimilar concept be applied to monoclonals? Regul Rapp. 2011;8(1):11–4.
Guidance for industry: good pharmacovigilance practices and pharmacoepidemiologic assessment. Rockville, MD: US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research; 2005.
European Medicines Agency. Committee for Medicinal Products For Human Use (CHMP). Guideline on the Investigation of Bioequivalence. 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed 30 March 2016.
Nick C. The US biosimilars act challenges facing regulatory approval. Pharm Med. 2012;26(3):145–52.
Cortes J, Curigliano G, Dieras V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat. 2014;144(2):233–9.
Cortazar P, Geyer CE Jr. Pathological complete response in neoadjuvant treatment of breast cancer. Ann Surg Oncol. 2015;22(5):1441–6.
Jackisch C, Scappaticci FA, Heinzmann D, Bisordi F, Schreitmuller T, Minckwitz G, et al. Neoadjuvant breast cancer treatment as a sensitive setting for trastuzumab biosimilar development and extrapolation. Future Oncol. 2015;11(1):61–71.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–75.
Schellekens H. When biotech proteins go off-patent. Trends Biotechnol. 2004;22(8):406–10.
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–6.
European Medicines Agency. Guideline on similar biological medicinal products. London: European Medicines Agency; 2004.
World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). Geneva: WHO; 2009. http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed 30 March 2016.
Biologics Price Competition and Innovation Act. Title VII—improving access to innovative medical therapies. Subtitle A—Biologics Price Competition and Innovation. Sec. 7002. Approval pathway for biosimilar biological products. 2009. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf. Accessed 30 March 2016.
US Food and Drug Administration. Guidance for industry: draft guidance: considerations in demonstrating interchangeability with a reference product. 2017. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed 20 Feb 2017.
US Food and Drug Administration. Guidance for industry: nonproprietary naming of biological products. 2017. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Accessed 20 Feb 2017.
Declerck PJ, Simoens S. A European perspective on the market accessibility of biosimilars. Biosimilars. 2012;2:33–40.
Grabowski H, Guha R, Salgado M. Biosimilar competition: lessons from Europe. Nat Rev Drug Discov. 2014;13(2):99–100.
Dylst P, Vulto A, Simoens S. Barriers to the uptake of biosimilars and possible solutions: a Belgian case study. Pharmacoeconomics. 2014;32(7):681–91.
Bocquet F, Loubiere A, Fusier I, Cordonnier AL, Paubel P. Competition between biosimilars and patented biologics: learning from European and Japanese experience. Pharmacoeconomics. 2016;34(11):1173–86.
Bocquet F, Paubel P. First monoclonal antibody biosimilars: tackling the challenge of substitution. J Med Econ. 2016;19(6):645–7.
European Medicines Agency. European public assessment reports. 2017. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124. Accessed 13 March 2017.
US Food and Drug Administration. Drugs@FDA: FDA approved drug products. 2017. http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=BasicSearch.process. Accessed 13 March 2017.
Acknowledgements
Editing support funded by Amgen Inc. was provided by Complete Healthcare Communications, LLC, an ICON plc company, Chadds Ford, PA, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
Richard Markus, Jennifer Liu, Monica Ramchandani, Diana Landa, Teresa Born, and Primal Kaur are employees of Amgen Inc. and own Amgen stock.
Funding
Funded by Amgen Inc.
Research Involving Human Participants and/or Animals
Not applicable.
Informed Consent
Not applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Markus, R., Liu, J., Ramchandani, M. et al. Developing the Totality of Evidence for Biosimilars: Regulatory Considerations and Building Confidence for the Healthcare Community. BioDrugs 31, 175–187 (2017). https://doi.org/10.1007/s40259-017-0218-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-017-0218-5