
Abstract
Psychosis occurs in approximately half of patients with Alzheimer disease (AD with psychosis, AD + P). AD + P patients have more rapid cognitive decline, greater behavioral symptoms, and higher mortality than do AD patients without psychosis. Studies in three independent cohorts have shown that psychosis in AD aggregates in families, with estimated heritability of 29.5–60.8 %. These findings have motivated studies to investigate and uncover the genes responsible for the development of psychosis, with the ultimate goal of identifying potential biologic mechanisms that may serve as leads to specific therapies. Linkage analyses have implicated loci on chromosomes 2, 6–8, 15, and 21 with AD + P. Association studies on apolipoprotein E do not support it as a risk gene for psychosis in AD. No other candidate genes, such as neurodegenerative and monoamine genes, show conclusive evidence of association with AD + P. However, a recent genome-wide association study has produced some promising leads, including among them genes that have been associated with schizophrenia. This review summarizes the current knowledge on the genetic basis of AD + P.
Similar content being viewed by others


Background
A subgroup of late-onset Alzheimer disease (AD) patients develops psychosis during progression of the disease (AD with psychosis, AD + P). Psychotic symptoms in AD are typically defined by the presence of delusions and hallucinations. Psychosis in AD subjects has been reported to range in prevalence from 12.2 to 74.1 % (median 41 %) and have a cumulative incidence approximating 50 % [1•]. The variability in reported prevalence may be explained by the fact that the development of AD + P is dependent on the stage of the disease, with low prevalence in the prodromal stage, a large increase in occurrence during early AD and highest prevalence in the middle disease stages [2, 3]. Numerous studies have shown that greater cognitive impairment is the most consistent correlate of AD + P compared to AD patients without psychosis [4]. With estimates of more than 13 million people affected with AD by the year 2050, AD + P may soon be the second most common psychotic disorder (schizophrenia being first) in the US [5].
The presence of psychotic symptoms in AD patients has a negative impact on the patient, family, and caregiver. AD + P patients often have other psychiatric and behavioral disturbances, the most frequent and troublesome of which are agitation [6] as well as verbal and physical aggression [7, 8]. Overall, AD + P leads to greater distress for family and caregivers [9], greater rates of institutionalization [10–13], and worse general health for the patient [14], with increased mortality [15, 16] compared to patients with AD − P.
Treatment of psychosis in AD patients with typical and atypical antipsychotics has been suboptimal. Their efficacy is low, and they have serious side effects for this population [17–19]. These drugs were developed for treatment of psychotic symptoms in a population without co-occurring dementia, so their biological specificity in AD + P patients may be poor. It is, therefore, important to determine the pathogenesis of psychosis in AD so that better pharmaceutical treatments can be developed for this subgroup of patients. During the past 10 years, substantial information from brain imaging, neuropathological, clinical, and genetic studies has begun to accrue regarding the neurobiology of AD + P [1•]. This review summarizes research findings regarding the genetic basis of psychosis in AD.
Familial Aggregation and Heritability
Several studies have examined whether psychosis in AD (AD + P) runs in families. An early study that examined familial aggregation in sibling pairs diagnosed with AD found that the frequency of psychosis in these patients was 0.41 [20]. The pair-wise concordance for psychosis was 0.21, which was modestly higher than expected by chance alone, 0.17 (Table 1•).
A larger study was done by Sweet et al. [21••] in which they looked at a family cohort in the National Institute of Mental Health (NIMH) AD Genetics Initiative. All probands and their siblings were diagnosed with AD and were tested for familial aggregation of AD + P. Of the 371 probands and their 461 siblings in the study, 75.5 % of the probands were positive for psychosis. They found a significant association between psychosis in the proband and the occurrence of AD + P in their siblings (Table 1). Similar results were obtained when covariates for age, age-of-onset, and the presence of extrapyramidal symptoms were accounted for. Interestingly, when a more restrictive definition of AD + P requiring multiple psychotic symptoms to be present over time was used in the model, the familial aggregation was strengthened.
Following this study, which showed that psychosis in AD aggregates in families, Bacanu et al. [22•] then employed statistical modeling to estimate the heritability of psychosis among 826 of the individuals from their prior report. The heritability of an occurrence of a single psychotic symptom was modest (29.5 %, p = 0.04), but when heritability was assessed using the more restrictive definition of psychosis requiring multiple or recurrent symptoms, it increased to 60.8 %, p = 0.004. The odds ratio (OR) for at least one psychotic symptom was 2.37 (1.45–3.87), and it increased to 5.42 (2.62–10.43) for multiple psychotic symptoms [22•] (Table 1).
Hollingworth et al. [23•] expanded the work of Sweet et al. by combining the data of the NIMH cohort with families recruited from the UK. A significant association existed between the psychosis status of the probands and the occurrence of AD + P in family members in both the NMIH and UK samples. The OR in the NIMH sample for the development of AD + P in siblings of probands with AD + P was 3.2 (2.05–4.99). Similar findings were obtained in the UK sample with an OR of 4.17 (1.67–10.44). For the combined sample of NIMH and UK families, the OR was 3.38 (2.27–5.05) (Table 1).
Sweet et al. [3] confirmed previous aggregation studies by looking at families in which multiple members were affected with late-onset AD and were part of the National Institute on Aging Late Onset AD Family Study. The association of psychosis in the proband with AD + P in other family members was highly significant (χ 2 = 15.8, df = 4, p = 0.003; Table 1).
In summary, studies of three independent cohorts have found evidence of significant familial aggregation of psychosis in AD. Study of a fourth cohort found suggestive evidence of the same. The estimated heritability of AD + P, when defined by multiple and/or recurrent psychotic symptoms, was 60.8 %. Taken as a whole, these findings provide compelling support for the hypothesis that psychosis in late-onset AD has a genetic basis and a firm rationale for studies exploring the underlying genetic associations.
Genetic Linkage Studies
With the identification that psychosis in AD aggregates in families, several studies were done to identify chromosomal loci that might be linked to and predispose AD patients to the development of psychosis. The first linkage study of AD + P found significant linkage on chromosome 2p and suggestive linkage on chromosomes 6 and 21 [24•]. Hollingworth et al. [23•] examined linkage using the combined NIMH and UK cohorts described earlier. In the NIMH sample, significant linkage was found on chromosomes 7 and 15. They also found suggestive evidence of linkage to loci on chromosomes 6 and 21; however, these loci were no longer significant after the inclusion of the apolipoprotein E (APOE) genotype as a covariate.
Neuregulin-1 (NRG1) on chromosome 8 is a gene of interest in psychosis because both linkage and association have been found for NRG1 in patients with schizophrenia [25]. A study examining 437 families from the NIMH AD Genetics Initiative found significant linkage for NRG1 and AD + P [26•]. Go et al. [26•] then analyzed four specific single nuclear polymorphisms (SNPs) within NRG1 for linkage and association with AD + P. Three of these SNPS were part of the Icelandic haplotype reported to be associated with risk for schizophrenia (SNP8NRG221533, SNP8NRG243177, SNP8NRG2419) [27]; the fourth was an exonic SNP (rs392499). In single SNP analyses, only rs392499 showed significant association with AD + P.
The previous studies that found significant linkage with psychosis to loci on chromosomes 2, 6–8, and 15 utilized a definition of psychosis that included the occurrence of either delusions or hallucinations. A study by Avramopoulos et al. [28] examined linkage separately for delusions and hallucinations. They found a region on chromosome 14 that was linked to AD patients without hallucinations. The linked region was close to, but independent of, the PSEN1 locus. They also found linkage of chromosome 2 with delusions in AD patients.
Genome-Wide Association Studies (GWASs)
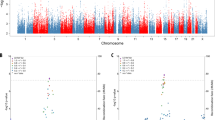
The first GWA analysis of AD + P was recently reported [29••], combining meta-analytically three AD GWA data sets [30–32]. The final analyzed sample included 1,299 cases with AD + P, 735 with AD − P and 5,659 controls unaffected by AD. After imputation, 1,882,172 SNPs were evaluated in the contrast of AD + P versus AD − P. The AD + P versus control contrast included 1,847,262 SNPs.
The results for the AD + P versus AD − P and AD + P versus control analysis are shown in Table 2. Among the most significant SNPs in the AD + P versus AD − P analysis was rs3764640 in serine/threonine kinase 11, STK11 [29••]. STK11 deletions are known to cause Peutz-Jeghers syndrome. However, a case with an unusually large STK11 deletion has been described in which Peutz-Jeghers syndrome, mental retardation, and schizophrenia co-occurred [33]. Similarly, a genome-wide screen in siblings co-affected by schizophrenia found reduced copy numbers of STK11 in 3/18 individuals, significantly more often than in controls [34]. Finally, STK11, also known as liver kinase B1, is a necessary intermediate in amyloid beta (Aβ) precursor protein (APP) overexpression-induced tau phosphorylation [35, 36].
In the AD + P versus control analysis, the most significant intragenic SNP was rs4038131, an intronic SNP in visinin-like 1 (VSNL1). rs4038131 also showed evidence of association with AD + P versus AD − P (OR 0.72, p = 1.84 × 10−2) [29••]. VSNL1 encodes the neuronal calcium sensor, visinin-like protein 1, Vilip1 [37]. Cerebrospinal fluid and plasma concentrations of Vilip1 are elevated in AD subjects in comparison to normal controls [38, 39] and to non-AD dementia subjects [39]. In early AD, elevated cerebrospinal fluid Vilip1 levels predict more rapid cognitive decline [40]. Of interest, expression of VSNL1 mRNA and Vilip1 protein is also reported to be altered in schizophrenia [41, 42].
In contrast to the above findings, a number of loci recently identified as associated with risk of AD, including clusterin, phosphatidylinositol-binding clathrin assembly protein, complement receptor 1, bridging integrator 1, ATP-binding cassette transporter 7, membrane-spanning 4-domain subfamily A, CD2-associated protein, CD33, and ephrin type-A receptor 1 were not associated with AD + P when compared to AD − P cases. Similarly, APOE/TOMM40 (translocase of outer mitochondrial membrane 40 homolog) SNPs were not associated with AD + P when compared to AD − P [29••].
Finally, prior GWASs had identified a number of loci with risk for schizophrenia and bipolar illness [43–49]. Hollingworth et al. [29••] sought to determine whether these SNPs might share an association with psychosis in AD. In the same GWAS cohorts described above, we tested 11 SNPs that had genome-wide evidence for association with schizophrenia or bipolar illness. Individually, none of the SNPs had an association with psychosis in AD; however, there was a trend toward association when all SNPs were grouped (combined p = 0.109).
Candidate Gene Studies
Prior to the advent of GWA, a large body of research was reported assessing the association of candidate genes with AD + P. The majority of such studies have focused understandably on the ∈4 allele of APOE. A large effort has also examined the monoamine neurotransmitter systems, namely serotonin and dopamine. With some limited exceptions, these individual studies have been characterized by small numbers of genetic variants assessed in any given study and small sample sizes, precluding any firm conclusions. Because candidate gene studies of APOE, serotonin system genes, and dopamine system genes have recently been comprehensively reviewed elsewhere [1•, 4], only a brief summary of these findings are presented below.
Association of APOE
The APOE ∈4 allele is a strong risk factor for the development of late-onset AD; therefore, candidate gene studies investigated its possible role in the development of psychosis in AD. In a review of 22 studies examining the association of APOE ∈4 with psychosis, 9 found a significant association, but the nature of the association (genotype, carrier status, allele frequency, etc.) differed across studies [4]. It was unclear whether the contrasting results reflected a lack of true association or may have arisen because of heterogeneity among the sample sizes, patient populations, and diagnostic criteria used. To attempt to address the issue of possible heterogeneity, DeMichele-Sweet et al. [50] examined the association of APOE ∈4 with AD + P in the National Alzheimer’s Disease Coordinating Center data set, which provided a large sample (N = 2,317) with uniform diagnostic criteria and measurement of psychosis. No association was found between psychosis and APOE ∈4 carrier status or APOE ∈4 allele number (Table 3). Since psychosis may not manifest until later in the course of disease, analyses were restricted to subjects who had reached at least a mild to moderate stage of illness (Clinical Dementia Rating Scale score ≥1, N = 1,941). In this follow-up, no association of APOE ∈4 carrier status and APOE ∈4 allele number with psychosis remained (Table 3).
Of note, a poly-T repeat sequence polymorphism in TOMM40, a locus in linkage disequilibrium with APOE, has been suggested to underlie the biological effects associated with genetic variation in APOE [51]. A recent study explored whether the variable associations of APOE with AD + P might result from an association with the TOMM40 repeat sequence. However, no association was found between the poly-T repeat length and psychosis [52].
Association of Monoamine System Genes
Genetic variations in serotonin receptors and transporters have been tested for association with AD + P because of the importance of the serotonin system in regulating central nervous system functions, including its involvement in psychiatric disorders [53, 54] and its altered levels in brains of AD subjects [55–57]. As with APOE, associations of polymorphisms and allele frequencies of serotonin receptors and transporters with psychosis in AD have been contradictory and do not generally support any clear associations (see Table S2 in [1•] for details).
Genetic variation in dopamine receptors has been of interest in association studies with AD + P because antipsychotic agents target these receptors [58]. The dopamine transporter gene has similarly been studied because of its role in regulating synaptic dopamine. However, as for serotonin system genes, results for dopamine system genetic variants have been inconclusive (see Table S2 in [1] for details). Similarly, the catechol-O-methyltransferase gene (COMT), which codes for an enzyme that inactivates dopamine, was evaluated in AD + P after several studies reported SNPs in COMT to be associated with schizophrenia [59]. However, these too yielded inconclusive results.
Association of Neurodegenerative Pathway Genes
The greatest association of cognitive impairment in subjects with AD is loss of synapses across the neocortical regions [60, 61]. Subjects with AD + P also have a more rapid cognitive decline than subjects without psychosis [1•], and this decline is associated with increased synaptic disruption across multiple neocortical regions in subjects with AD + P [62]. Soluble Aβ protein directly leads to synapse loss [63–66], and it may also increase aggregation of microtubule-associated protein tau, MAPT [66], which itself can contribute to loss of synapses [67]. Therefore, genes that regulate Aβ and MAPT represent potential candidates for association with AD + P.
DeMichele-Sweet et al. [68] comprehensively evaluated the APP and MAPT genes for association with AD + P in a cohort of 867 well-characterized subjects. They also evaluated sortilin-related receptor (SORL1), an AD-risk gene that impacts Aβ metabolism and correlates with measures of synaptic markers [69–71], and β-site amyloid precursor protein cleaving enzyme (BACE1), an enzyme involved in the conversion of APP to Aβ [72]. There was no evidence of an association of APP, SORL1, BACE1, and MAPT with the occurrence of psychosis in AD [68].
Miscellaneous Associations
The α7 nicotinic acetylcholine receptor is encoded by CHRNA7 on chromosome 15 and has been reported to demonstrate significant linkage and association in schizophrenia [73, 74]. An initial study by Carson et al. [75] looked at whether this gene is associated with psychosis in AD. Analyzing 14 SNPs in this gene in a group of 409 probable AD patients of Northern Irish descent, they found a significant association between a single SNP (rs6494223) and delusions in AD (p = 0.017) with risk conveyed by the T allele (OR = 1.63, CI = 1.22–2.17). This finding has yet to be replicated in other AD populations.
A polymorphism of the interleukin 1β gene promoter was studied in a population of 424 patients diagnosed with possible/probable AD in the UK [76]. The CC genotype frequency was significantly higher in patients with delusions (χ 2 = 2.69, p = 0.002), with hallucinations (χ 2 = 6.27, p = 0.043), and with both delusions and hallucinations (χ 2 = 9.9, p = 0.007). The frequency of the C allele was also significantly higher in patients with delusions (χ 2 = 4.86, OR = 1.49, CI = 1.02–1.94, p = 0.028), with hallucinations (χ2 = 5.95, OR = 1.6, CI = 1.08–2.39, p = 0.014), and with both (χ 2 = 3.91, OR = 1.62, CI = 0.98–2.70, p = 0.048). Although this provides intriguing evidence for a potential role of inflammation in psychosis risk in AD, independent confirmation of these findings is pending.
Conclusion
Psychosis occurs in a subset of patients with AD, in whom it is associated with a more aggressive cognitive deterioration and worse outcomes. There is now evidence from three independent replications that psychosis in AD aggregates within families and thus is likely to result, in part, from effects of genetic variation. At present, there is no gene in which genetic variation can unequivocally be stated to associate with the risk of psychosis in AD, although several promising leads from a recent GWAS have been identified. In contrast, substantial evidence supports the conclusion that the APOE locus can be excluded from an association with AD + P. Although based on much more limited evaluation, current evidence similarly suggests that other genetic variants that increase the risk for AD do not increase the risk for psychosis in AD. In contrast, although also derived from limited data, current evidence is consistent with the hypothesis that there is some overlap of genetic risk for psychosis in AD with that for schizophrenia.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Murray PS, Kumar S, DeMichele-Sweet MA, Sweet RA. Psychosis in Alzheimer’s disease. Biol Psychiatry. (2013, in press). Provides a comprehensive review of genetic, neuroimaging, and neuropathology of psychosis in Alzheimer disease.
Drevets WC, Rubin EH. Psychotic symptoms and the longitudinal course of senile dementia of the Alzheimer type. Biol Psychiatry. 1989;25:39–48.
Sweet RA, Bennett DA, Graff-Radford NR, Mayeux R. Assessment and familial aggregation of psychosis in Alzheimer’s disease from the National Institute on Aging Late Onset Alzheimer’s Disease Family Study. Brain. 2010;133:1155–62.
DeMichele-Sweet MA, Sweet RA. Genetics of psychosis in Alzheimer’s disease: a review. J Alzheimers Dis. 2010;19:761–80.
Alzheimers Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9:208–45.
Gilley DW, Whalen ME, Wilson RS, Bennett DA. Hallucinations and associated factors in Alzheimer’s disease. J Neuropsychiatry. 1991;3:371–6.
Gilley DW, Wilson RS, Beckett LA, Evans DA. Psychotic symptoms and physically aggressive behavior in Alzheimer’s disease. J Am Geriatr Soc. 1997;45:1074–9.
Sweet RA, Pollock BG, Sukonick DL, Mulsant BH, Rosen J, Klunk WE, Kastango KB, DeKosky ST. The 5-HTTPR polymorphism confers liability to a combined phenotype of psychotic and aggressive behavior in Alzheimer’s disease. Int Psychogeriatr. 2001;13:401–9.
Kaufer DI, Cummings JL, Christine D, Bray T, Castellon S, Masterman D, MacMillan A, Ketchel P, DeKosky ST. Assessing the impact of neuropsychiatric symptoms in Alzheimer’s disease: the Neuropsychiatric Inventory Caregiver Distress Scale. J Am Geriatr Soc. 1998;46:210–5.
Rabins PV, Mace NL, Lucas MJ. The impact of dementia on the family. JAMA. 1982;248:333–5.
Lopez OL, Wisniewski SR, Becker JT, Boller F, DeKosky ST. Psychiatric medication and abnormal behavior as predictors of progression in probable Alzheimer disease. Arch Neurol. 1999;56:1266–72.
Magni E, Binetti G, Bianchetti A, Trabucchi M. Risk of mortality and institutionalization in demented patients with delusions. J Geriatr Psychiatry Neurol. 1996;9:123–6.
Cummings JL, Diaz C, Levy M, Binetti G, Litvan II. Neuropsychiatric syndromes in neurodegenerative disease: frequency and significance. Semin Clin Neuropsychiatry. 1996;1:241–7.
Bassiony MM, Steinberg M, Rosenblatt A, Baker A, Lyketsos CG. Delusions and hallucinations in Alzheimer’s disease: prevalence and clinical correlates. Int J Geriatr Psychiatry. 2000;15:99–107.
Wilson RS, Krueger KR, Kamenetsky JM, Tang Y, Gilley DW, Bennett DA, Evans DA. Hallucinations and mortality in Alzheimer disease. Am J Geriatr Psychiatry. 2005;13:984–90.
Lopez OL, Becker JT, Chang YF, Sweet RA, Aizenstein H, Snitz B, Saxton J, McDade E, Kamboh MI, DeKosky ST, Reynolds CF III, Klunk WE. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170:1051–8.
Huybrechts KF, Gerhard T, Crystal S, Olfson M, Avorn J, Levin R, Lucas JA, Schneeweiss S. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977.
Schneider LS, Pollock VE, Lyness SA. A metaanalysis of controlled trials of neuroleptic treatment in dementia. J Am Geriatr Soc. 1990;38:553–63.
Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. 2006;14:191–210.
Tunstall N, Owen MJ, Williams J, Rice F, Carty S, Lillystone S, Fraser L, Kehoe P, Neill D, Rudrasingham V, Sham P, Lovestone S. Familial influence on variation in age of onset and behavioural phenotype in Alzheimer’s disease. Br J Psychiatry. 2000;176:156–9.
•• Sweet RA, Nimgaonkar VL, Devlin B, Lopez OL, DeKosky ST. Increased familial risk of the psychotic phenotype of Alzheimer disease. Neurology. 2002;58:907–11. One of the first studies of familial aggregation of psychosis in siblings of probands with Alzheimer disease and psychosis.
• Bacanu SA, Devlin B, Chowdari KV, DeKosky ST, Nimgaonkar VL, Sweet RA. Heritability of psychosis in Alzheimer disease. Am J Geriatr Psychiatry. 2005;13:624–7. Initial study that used statistical modeling to estimate the heritability of psychosis in Alzheimer patients. Results showed that heritability was 60.8 %.
• Hollingworth P, Hamshere ML, Holmans PA, O’Donovan MC, Sims R, Powell J, Lovestone S, Myers A, Devrieze FW, Hardy J, Goate A, Owen M, Williams J. Increased familial risk and genomewide significant linkage for Alzheimer’s disease with psychosis. Am J Med Genet B. 2007;144B:841–8. Another aggregation study of psychosis in Alzheimer disease. It employed a larger number of subjects from two different data sets. It also found a significant association between the psychosis status of the proband and the occurence of psychosis in family members with Alzheimer disease. This study also found significant linkage of chromosomes 7 and 15 with psychosis in Alzheimer disease.
• Bacanu SA, Devlin B, Chowdari KV, DeKosky ST, Nimgaonkar VL, Sweet RA. Linkage analysis of Alzheimer disease with psychosis. Neurology. 2002;59:118–20. The first study to identify chromosomal loci that might be linked to and predispose Alzheimer disease patients to develop psychosis. Significant linkage was found on chromosome 2p and suggestive linkage was found on chromosomes 6 and 21.
Harrison PJ, Law AJ. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006;60:132–40.
• Go RC, Perry RT, Wiener H, Bassett SS, Blacker D, Devlin B, Sweet RA. Neuregulin-1 polymorphism in late onset Alzheimer’s disease families with psychoses. Am J Med Genet B. 2005;139B:28–32. Linkage analysis of 4 specific SNPS with NRG1 with psychosis in Alzheimer disease.
Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, Hjaltason O, Birgisdottir B, Jonsson H, Gudnadottir VG, Gudmundsdottir E, Bjornsson A, Ingvarsson B, Ingason A, Sigfusson S, Hardardottir H, Harvey RP, Lai D, Zhou M, Brunner D, Mutel V, Gonzalo A, Lemke G, Sainz J, Johannesson G, Andresson T, Gudbjartsson D, Manolescu A, Frigge ML, Gurney ME, Kong A, Gulcher JR, Petursson H, Stefansson K. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–92.
Avramopoulos D, Fallin MD, Bassett SS. Linkage to chromosome 14q in Alzheimer’s disease (AD) patients without psychotic symptoms. Am J Med Genet B. 2005;132B:9–13.
•• Hollingworth P, Sweet R, Sims R, Harold D, Russo G, Abraham R, Stretton A, Jones N, Gerrish A, Chapman J, Ivanov D, Moskvina V, Lovestone S, Priotsi P, Lupton M, Brayne C, Gill M, Lawlor B, Lynch A, Craig D, McGuinness B, Johnston J, Holmes C, Livingston G, Bass NJ, Gurling H, McQuillin A, Holmans P, Jones L, Devlin B, Klei L, Barmada MM, Demirci FY, DeKosky ST, Lopez OL, Passmore P, Owen MJ, O’Donovan MC, Mayeux R, Kamboh MI, Williams J. Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol Psychiatry. 2012;17:1316–27. This study was the first genome-wide association (GWA) analysis of psychosis in Alzheimer disease (AD). It meta-analytically combined three AD GWA datasets. The final analyzed sample included 1299 AD cases with psychosis, 735 AD subjects without psychosis, and 5659 controls unaffected by AD. After imputation, 1,882,172 single nuclear polymorphisms (SNPs) were evaluated in the contrast of AD + P vs AD − P. The AD + P vs Control contrast included 1,847,262 SNPs.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93.
Wijsman EM, Pankratz ND, Choi Y, Rothstein JH, Faber KM, Cheng R, Lee JH, Bird TD, Bennett DA, Diaz-Arrastia R, Goate AM, Farlow M, Ghetti B, Sweet RA, Foroud TM, Mayeux R. Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011;7:e1001308.
Kamboh MI, Demirci FY, Wang X, Minster RL, Carrasquillo MM, Pankratz VS, Younkin SG, Saykin AJ, Jun G, Baldwin C, Logue MW, Buros J, Farrer L, Pericak-Vance MA, Haines JL, Sweet RA, Ganguli M, Feingold E, DeKosky ST, Lopez OL, Barmada MM. Genome-wide association study of Alzheimer’s disease. Transl Psychiatry. 2012;2:e117.
Kam M, Massare J, Gallinger S, Kinzie J, Weaver D, Dingell JD, Esufali S, Bapat B, Tobi M. Peutz–Jeghers syndrome diagnosed in a schizophrenic patient with a large deletion in the STK11 gene. Dig Dis Sci. 2006;51:1567–70.
Lee CH, Liu CM, Wen CC, Chang SM, Hwu HG. Genetic copy number variants in sib pairs both affected with schizophrenia. J Biomed Sci. 2010;17:2.
Kojima Y, Miyoshi H, Clevers HC, Oshima M, Aoki M, Taketo MM. Suppression of tubulin polymerization by the LKB1-microtubule-associated protein/microtubule affinity-regulating kinase signaling. J Biol Chem. 2007;282:23532–40.
Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci. 2007;27:574–81.
Amici M, Doherty A, Jo J, Jane D, Cho K, Collingridge G, Dargan S. Neuronal calcium sensors and synaptic plasticity. Biochem Soc Trans. 2009;37:1359–63.
Lee JM, Blennow K, Andreasen N, Laterza O, Modur V, Olander J, Gao F, Ohlendorf M, Ladenson JH. The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin Chem. 2008;54:1617–23.
Tarawneh R, D’Angelo G, Macy E, Xiong C, Carter D, Cairns NJ, Fagan AM, Head D, Mintun MA, Ladenson JH, Lee JM, Morris JC, Holtzman DM. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011;70:274–85.
Tarawneh R, Lee JM, Ladenson JH, Morris JC, Holtzman DM. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology. 2012;78:709–19.
Beveridge NJ, Tooney PA, Carroll AP, Gardiner E, Bowden N, Scott RJ, Tran N, Dedova I, Cairns MJ. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet. 2008;17:1156–68.
Bernstein HG, Braunewell KH, Spilker C, Danos P, Baumann B, Funke S, Diekmann S, Gundelfinger ED, Bogerts B. Hippocampal expression of the calcium sensor protein visinin-like protein-1 in schizophrenia. Neuroreport. 2002;13:393–6.
O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, Dwyer S, Holmans P, Marchini JL, Spencer CC, Howie B, Leung HT, Hartmann AM, Moller HJ, Morris DW, Shi Y, Feng G, Hoffmann P, Propping P, Vasilescu C, Maier W, Rietschel M, Zammit S, Schumacher J, Quinn EM, Schulze TG, Williams NM, Giegling I, Iwata N, Ikeda M, Darvasi A, Shifman S, He L, Duan J, Sanders AR, Levinson DF, Gejman PV, Cichon S, Nothen MM, Gill M, Corvin A, Rujescu D, Kirov G, Owen MJ, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Cloninger CR. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–5.
O’Donovan MC, Craddock NJ, Owen MJ. Genetics of psychosis; insights from views across the genome. Hum Genet. 2009;126:3–12.
Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–7.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de Frutos R, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St Clair D, Goldstein DB, Stefansson K, Collier DA. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7.
Cichon S, Muhleisen TW, Degenhardt FA, Mattheisen M, Miro X, Strohmaier J, Steffens M, Meesters C, Herms S, Weingarten M, Priebe L, Haenisch B, Alexander M, Vollmer J, Breuer R, Schmal C, Tessmann P, Moebus S, Wichmann HE, Schreiber S, Muller-Myhsok B, Lucae S, Jamain S, Leboyer M, Bellivier F, Etain B, Henry C, Kahn JP, Heath S, Hamshere M, O’Donovan MC, Owen MJ, Craddock N, Schwarz M, Vedder H, Kammerer-Ciernioch J, Reif A, Sasse J, Bauer M, Hautzinger M, Wright A, Mitchell PB, Schofield PR, Montgomery GW, Medland SE, Gordon SD, Martin NG, Gustafsson O, Andreassen O, Djurovic S, Sigurdsson E, Steinberg S, Stefansson H, Stefansson K, Kapur-Pojskic L, Oruc L, Rivas F, Mayoral F, Chuchalin A, Babadjanova G, Tiganov AS, Pantelejeva G, Abramova LI, Grigoroiu-Serbanescu M, Diaconu CC, Czerski PM, Hauser J, Zimmer A, Lathrop M, Schulze TG, Wienker TF, Schumacher J, Maier W, Propping P, Rietschel M, Nothen MM. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet. 2011;88:372–81.
Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, Smoller JW, Grozeva D, Stone J, Nikolov I, Chambert K, Hamshere ML, Nimgaonkar VL, Moskvina V, Thase ME, Caesar S, Sachs GS, Franklin J, Gordon-Smith K, Ardlie KG, Gabriel SB, Fraser C, Blumenstiel B, Defelice M, Breen G, Gill M, Morris DW, Elkin A, Muir WJ, McGhee KA, Williamson R, MacIntyre DJ, MacLean AW, St Clair D, Robinson M, Van Beck M, Pereira AC, Kandaswamy R, McQuillin A, Collier DA, Bass NJ, Young AH, Lawrence J, Ferrier IN, Anjorin A, Farmer A, Curtis D, Scolnick EM, McGuffin P, Daly MJ, Corvin AP, Holmans PA, Blackwood DH, Gurling HM, Owen MJ, Purcell SM, Sklar P, Craddock N, Welcome Trust Case Control Consortium. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–8.
McMahon FJ, Akula N, Schulze TG, Muglia P, Tozzi F, Detera-Wadleigh SD, Steele CJ, Breuer R, Strohmaier J, Wendland JR, Mattheisen M, Muhleisen TW, Maier W, Nothen MM, Cichon S, Farmer A, Vincent JB, Holsboer F, Preisig M, Rietschel M, Bipolar Disorder Genome Study (BiGS) Consortium. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat Genet. 2010;42:128–31.
DeMichele-Sweet MA, Lopez OL, Sweet RA. Psychosis in Alzheimer’s disease in the national Alzheimer’s disease coordinating center uniform data set: clinical correlates and association with apolipoprotein e. Int J Alzheimers Dis. 2011;2011:926597.
Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10:375–84.
Chu SH, Roeder K, Ferrell RE, Devlin B, DeMichele-Sweet MA, Kamboh MI, Lopez OL, Sweet RA. TOMM40 poly-T repeat lengths, age of onset and psychosis risk in Alzheimer disease. Neurobiol Aging. 2011;32:2328–9.
Cross AJ. Serotonin in Alzheimer-type dementia and other dementing illnesses. Ann NY Acad Sci. 1990;600:405–17.
Meltzer CC, Smith G, DeKosky ST, Pollock BG, Mathis CA, Moore RY, Kupfer DJ, Reynolds CF III. Serotonin in aging, late-life depression, and Alzheimer’s disease: the emerging role of functional imaging. Neuropsychopharmacology. 1998;18:407–30.
Bowen DM, Allen SJ, Benton JS, Goodhardt MJ, Haan EA, Palmer AM, Sims NR, Smith CC, Spillane JA, Esiri MM, Neary D, Snowdon JS, Wilcock GK, Davison AN. Biochemical assessment of serotonergic and cholinergic dysfunction and cerebral atrophy in Alzheimer’s disease. J Neurochem. 1983;41:266–72.
Cheng AV, Ferrier IN, Morris CM, Jabeen S, Sahgal A, McKeith IG, Edwardson JA, Perry RH, Perry EK. Cortical serotonin-S2 receptor binding in Lewy body dementia, Alzheimer’s and Parkinson’s diseases. J Neurol Sci. 1991;106:50–5.
Tejani-Butt SM, Yang J, Pawlyk AC. Altered serotonin transporter sites in Alzheimer’s disease raphe and hippocampus. Neuroreport. 1995;6:1207–10.
Seeman P. Dopamine receptor sequences: therapeutic levels of neuroleptics occupy D2 receptors, clozapine occupies D4. Neuropsychopharmacology. 1992;7:261–84.
Shifman S, Bronstein MSM, Pisante-Shalom A, Lev-Lehman E, Weizman A, Reznik I, Spivak B, Grisaru N, Karp L, Schiffer R, Kotler M, Strous RD, Swartz-Vanetik M, Knobler HY, Shinar E, Beckmann JS, Yakir B, Risch N, Zak NB, Darvasi A. A highly significant association between a COMT haplotype and schizophrenia. Am J Hum Genet. 2002;71:1296–302.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80.
Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–46.
Sweet RA, Panchalingam K, Pettegrew JW, McClure RJ, Hamilton RL, Lopez OL, Kaufer DI, DeKosky ST, Klunk WE. Psychosis in Alzheimer disease: postmortem magnetic resonance spectroscopy evidence of excess neuronal and membrane phospholipid pathology. Neurobiol Aging. 2002;23:547–53.
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–62.
Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–7.
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91.
Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem. 2007;101:1172–84.
Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schonig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007;282:31755–65.
DeMichele-Sweet MA, Klei L, Devlin B, Ferrell RE, Weamer EA, Emanuel JE, Lopez OL, Sweet RA. No association of psychosis in Alzheimer disease with neurodegenerative pathway genes. Neurobiol Aging. 2011;32:555.e9–11.
Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–77.
Mayeux R, George-Hyslop P. Brain traffic: subcellular transport of the amyloid precursor protein. Arch Neurol. 2009;66:433–4.
Grear KE, Ling IF, Simpson JF, Furman JL, Simmons CR, Peterson SL, Schmitt FA, Markesbery WR, Liu Q, Crook JE, Younkin SG, Bu G, Estus S. Expression of SORL1 and a novel SORL1 splice variant in normal and Alzheimers disease brain. Mol Neurodegener. 2009;4:46.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–41.
Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA. 1997;94:587–92.
Freedman R, Leonard S, Gault JM, Hopkins J, Cloninger CR, Kaufmann CA, Tsuang MT, Farone SV, Malaspina D, Svrakic DM, Sanders A, Gejman P. Linkage disequilibrium for schizophrenia at the chromosome 15q13-14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7). Am J Med Genet. 2001;105:20–2.
Carson R, Craig D, Hart D, Todd S, McGuinness B, Johnston JA, O’Neill FA, Ritchie CW, Passmore AP. Genetic variation in the alpha 7 nicotinic acetylcholine receptor is associated with delusional symptoms in Alzheimer’s disease. Neuromol Med. 2008;10:377–84.
Craig D, Hart DJ, McCool K, McIlroy SP, Passmore AP. The interleukin 1beta gene promoter polymorphism (−511) acts as a risk factor for psychosis in Alzheimer’s dementia. Ann Neurol. 2004;56:121–4.
Acknowledgments
This work was supported by Veterans Health Administration Grant BX000452 and NIH Grants AG05133 and AG027224. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Department of Veterans Affairs, or the US Government.
Disclosures
Mary Ann A. DeMichele-Sweet has no conflicts of interest or financial interests to declare. Robert A. Sweet has served as a consultant for Lilly, USA.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
DeMichele-Sweet, M.A.A., Sweet, R.A. Genetics of Psychosis in Alzheimer Disease. Curr Genet Med Rep 2, 30–38 (2014). https://doi.org/10.1007/s40142-014-0030-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-014-0030-1