
Abstract
Epigenetic regulation, such as DNA methylation and histone modification, is implicated in the aberrant changes in gene expression that occur during the progression of neurodegeneration. Many epigenetics-based drugs have been developed recently for the treatment of some neurodegenerative disorders, including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Here we review recent studies that highlight the role of epigenetic modifications in neurodegeneration, among them DNA methylation and demethylation and histone acetylation and deacetylation; we also explore the possibility of using epigenetics-based therapeutics to treat neurodegenerative disorders.
Similar content being viewed by others


Introduction
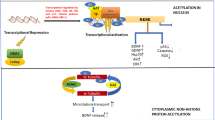
The term “epigenetics” encompasses all meiotically and mitotically heritable changes in gene expression that are not coded in the DNA sequence itself, and only alter phenotype without changing genotype [1]. Epigenetic modifications include DNA methylation, histone modifications, the action of small RNAs, and nucleosome positioning [2–4]. These modifications form a network of covalent alterations to DNA and histone proteins, which in turn interact with other cellular proteins, typically in multi-component mediator complexes, the end result being altered gene expression. The recognition of the role epigenetics plays in human disease began with oncology, but has now extended to other diseases, such as neurodevelopment and neurodegenerative disorders. Epigenetic interventions affect the compaction of chromatin and alter the binding of histones to promoter regions of their associated DNA surrounding the nucleosomal core, thereby influencing the accessibility of transcription factors and their ability to regulate gene expression.
There has been considerable progress in the development of epigenetic drugs for the treatment of neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Several drugs that target epigenetic alterations, including inhibitors of histone deacetylase (HDAC) and DNA methyltransferase (DNMT), are currently approved and available for clinical investigation in these disorders [5••]. In this review, we will discuss the role of epigenetic modifications in neurodegeneration, including DNA methylation and demethylation and histone acetylation and deacetylation; we also explore the possibility of using epigenetics-based therapeutics to treat neurodegenerative disorders.
Epigenetic Alterations in Neurodegeneration
DNA Methylation and Demethylation
DNA methylation, the most widely studied epigenetic mechanism, is catalyzed by a family of DNA methyltransferases (DNMTs) that transfer a methyl group from S-adenosylmethionine (SAM) to the fifth carbon of a cytosine residue to form 5mC. In mammals, there are five known members of the DNMT family: DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L (DNMT3-like), but only DNMT1, DNMT3a, and DNMT3b possess methyltransferase activity [4]. The catalytic members of the DNMT family are customarily classified into de novo DNMTs (DNMT3a and DNMT3b) and maintenance DNMTs (DNMT1). DNMT3a and DNMT3b can establish a new methylation pattern in unmodified DNA and are thus known as de novo DNMTs. These DNMTs are highly expressed in embryonic stem (ES) cells and downregulated in differentiated cells [6]. On the other hand, the maintenance DNMTs function during DNA replication to copy the DNA methylation pattern from the parental DNA strand onto the newly synthesized daughter strand [7•]. DNMT1 has a 30-fold to 40-fold preference for hemimethylated DNA, and also has de novo DNMT activity. DNMT1 is the most abundant DNMT in the cell and is transcribed mostly during the S-phase of the cell cycle. It is most often needed to methylate hemimethylated sites that are generated during semi-conservative DNA replication. In a cellular context, the affinity of DNMT1 for newly synthesized DNA is increased by its interaction with the DNA polymerase processing factor proliferating cell nuclear antigen (PCNA), ensuring localization to the replication fork [8]; the ubiquitin-like plant homeodomain and RING finger domain-containing protein 1 (UHRF1) could perform a similar function, tethering DNMT1 to hemimethylated DNA [9].
Most DNA methylation occurs on cytosines that precede a guanine nucleotide or CpG sites. It is essential for silencing retroviral elements, regulating tissue-specific gene expression, genomic imprinting, and X chromosome inactivation. Importantly, DNA methylation in different genomic regions may exert different influences on gene activities based on the underlying genetic sequence [7•]. The proteins that bind to and recognize 5-methylcytosines are the methyl-CpG binding domain (MBD) proteins: in mammals, these are MeCP2, MBD1, MBD2, MBD3, and MBD4. MeCP2 was the first of these proteins to be characterized [10]. Almost all MBD proteins are localized in highly methylated chromatin regions involved in silencing imprinted genes and in endoparasitic sequences [11], where they promote genomic stability and transcriptional repression.
DNA demethylation occurs via a series of chemical reactions that further modify 5mC. Several mechanisms of active DNA demethylation have been proposed. 5mC can be chemically modified at two sites, the amine group and the methyl group. Deamination of the amine to a carbonyl group by AID/APOBEC (activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme complex) effectively converts 5mC into thymine, thus creating a G/T mismatch and inducing the base excision repair (BER) pathway to correct the base. Overexpression of AID/APOBEC promotes DNA demethylation in zebrafish [12], whereas knockdown or knockout inhibits the DNA demethylation of various genes necessary for cellular reprogramming and development [13, 14]. Another active DNA demethylation mechanism could be mediated by the ten–eleven translocation (TET) enzymes TET1, TET2, and TET3. TET enzymes add a hydroxyl group onto the methyl group of 5mC to form 5hmC [15••, 16]. The mature brain contains significant 5hmC levels in multiple regions, ranging from 0.3 % to 0.7 %, which is approximately tenfold lower than the average abundance of 5mC [17]. Once 5hmC is formed, two separate mechanisms can convert 5hmC back into cytosine in mammals: iterative oxidation by TET enzymes continues to oxidize 5hmC first to 5-formyl-cytosine (5fC) and then to 5-carboxy-cytosine (5caC) [18]; alternatively, 5hmC is deaminated by AID/APOBEC to form 5-hydroxymethyl-uracil (5hmU) [19•]. 5fC, 5caC, and 5hmU could be further converted into unmodified cytosine.
Histone Modifications
Histone modifications are the other major epigenetic marks. DNA is packed in the nucleus of eukaryotic cells through its chromatin organization. The nucleosome, the fundamental unit of chromatin structure, consists of 146 base pairs of DNA wrapped around an octamer of histones made up of two copies of each of the core histones: H2A, H2B, H3, and H4 [20]. These proteins not only provide a solid structure, N-terminal regions of histones protrude from the nucleosome and are susceptible to interactions with other proteins. There are many types of post-translational modification of the residues at histone tails, including methylation of lysines and arginines, acetylation, phosphorylation, ubiquitination, SUMOylation, and ADP-ribosylation.
The post-translational modification of histones is often a dynamic and reversible process mediated by two antagonistic sets of enzymatic complexes that attach or remove the corresponding chemical groups in a site-specific manner. One of the most thoroughly studied modifications of histone tails is the acetylation at lysine residues, which is associated with transcriptional activation [20]. Histone acetyltransferases (HATs) carry out these modifications, which can be reverted by histone deacetylases (HDACs). HATs are evolutionarily conserved from yeast to humans, and are categorized into three main families, GNAT, MYST, and CBP/p300, according to the structure of the catalytic domains [20, 21]. Acetylated histones serve as epigenetic markers, which could recruit HATs and other bromodomain-containing proteins. Histone deacetylases (HDACs) remove acetyl groups from lysine/arginine residues in the amino-terminal tails of core histones and other proteins, thereby reversing the effects of the HATs [20, 21]. Mammalian HDACs fall into four main classes, classes 1–4, with class 1 and class 2 HDACs receiving the most attention in the nervous system [38]. Class 1 HDACs (HDACs 1, 2, 3, and 8) are homologs of the yeast HDAC RPD3, are constitutively nuclear proteins, and are widely expressed. Class 2 HDACs (HDACs 4, 5, 6, 7, 9, and 10) are homologs of yeast Hda1, are expressed in a tissue-specific and cell-specific manner, and are regulated, at least in part, by shuttling between the nucleus and cytoplasm, where they are stabilized by interactions with 14-3-3 proteins. Sequence specificity of HDAC action is acquired by recruitment of HDACs to specific genetic loci by repressors, co-repressors, and methyl-DNA binding proteins [38].
Other histone modifications also take place on lysines at their tails, which can be monomethylated, dimethylated, or trimethylated. The effect of each modification depends on both the identity of the modified residue and the extent of methylation. For example, methylation of histone H3 on lysines 4 and 36 (H3K4 and H3K36) is generally associated with an “open”euchromatin structure and transcriptional activation, whereas methylation of histone H3 on lysines 9 and 27 (H3K9 and H3K27) is generally associated with a“closed”heterochromatin structure and gene silencing [20, 22]. Methylation of histones is carried out by histone methyltransferases and demethylation by histone demethylases, such as the jumonji protein family [23].
Potential Targets for Epigenetic Therapeutic Interventions
DNA Methylation and DNMT Inhibitors
DNA methylation is a conserved epigenetic mechanism that is used to transduce environmental influences into enduring changes in neuronal function and circuitry [24, 25]. An effective epigenetic pharmacological strategy for selectively reducing the expression of genes may involve methylating cytosine bases in promoter regions of DNA by increasing endogenous levels of the methyl donor S-adenosylmethionine (SAM) that is used in DNA methylation reactions via exogenous administration of methionine, choline, folates, and vitamin B12 [25, 26]. The selectivity of the latter epigenetic therapeutic strategy depends on increasing DNA methylation, thereby interfering with the binding of transcription factors to promoter regions that contain a high guanine-cytosine content of islands of CpG-enriched dinucleotides. Increasing levels of SAM can also lead to binding of proteins that recognize methylated cytosine bases in enriched CpG islands in promoters and recruit enzymes with HDAC activity, leading to chromatin compaction as an alternative mechanism of gene repression [27].
DNMT inhibitors, also referred to as demethylating agents, have been under preclinical and clinical investigation for over 30 years. The most widely studied of these have been the nucleoside analogs 5-azacitidine (AZA) and decitabine (DAC). Because they are cytidine analogs, both agents are incorporated into DNA after activation to a triphosphate moiety. After formation of an irreversible complex with DNMT1, degradation of the enzyme occurs [28]. This prevents methylation of daughter DNA in CpG islands during DNA replication. In addition, AZA (but not DAC) is converted into a ribonucleoside moiety and is incorporated into RNA, interfering with protein translation. A Phase III randomized trial in patients with high-risk myelodysplastic syndrome (MDS) indicated a significant improvement in median overall survival with AZA versus conventional care regimens, including best supportive care or standard chemotherapy (24.5 months versus 15 months) [29]. Impressive disease responses were also seen with the use of DAC in a similar patient population [29].
Other DNMT inhibitors in an earlier phase of development include DAC analogs, such as SGI-110 and zebularine. When administered directly into the brain tissue of mice and rats, these and other DNMT inhibitors disrupt synaptic plasticity and hippocampal learning and memory, and they are powerful modulators of reward and addiction behaviors [30, 31•, 32, 33]; however, nucleoside analogs are thought to act primarily at sites of DNA synthesis and replication during the cell cycle, making them seem at first glance to be of little relevance to postmitotic neurons and glia. Notably, N-phthalyl-L-tryptophan (RG 108) and other drugs that interfere with sites at which DNMT is active independent of DNA replication still cause a robust impairment of hippocampal learning and memory [34, 35]. The potential of drugs that interfere with DNMT activity may go beyond these examples of synaptic and behavioral plasticity. In mice, for example, treatment with DNMT inhibitors can confer stroke protection after mild ischemia; furthermore, haploinsufficiency for Dnmt1 in mice is associated with smaller infarction volumes after acute ischemia and stroke [36, 37]. For neurological diseases, the few DNA methylation markers that have been identified so far require further validation in large-scale multicenter studies.
Histone Acetylation and HDAC Inhibitors
Histone acetylation is of great interest in the nervous system, due in part to our knowledge of the enzymatic machinery and signal transduction mechanisms that regulate this post-translational modification. Acetylation of core histones is catalyzed by transcriptional co-activators, such as CREB-binding protein (CBP), which possess histone acetyltransferase (HAT) activity [20, 21]. Histone acetylation remodels chromatin structure, thereby modulating transcription. Specificity of gene regulation is achieved via the recruitment of HATs by transcription factors to specific genetic loci, where they locally modify histones. Importantly, HATs interact with a large number of transcription factors and thus serve as crucial hubs, integrating the activity of multiple signaling cascades. Deacetylation of histone proteins shifts the balance toward chromatin condensation and thereby silences gene expression. Unlike HATs, HDACs have a rich structural diversity, which confers diversity of function and makes HDACs promising targets for drug discovery and therapeutic intervention.
HDAC inhibitors can be classified into four main chemical families: the short-chain fatty acids (e.g. sodium butyrate, phenylbutyrate, and valproic acid); the hydroxamic acids (e.g. trichostatin A and suberoylanilide hydroxamic acid (SAHA)); the epoxyketones (e.g. trapoxin); and the benzamides. Of these, the most widely studied are sodium butyrate, phenylbutyrate, trichostatin A, and SAHA; the butyrates are known to cross the blood-brain barrier [38, 39]. In the nervous system, the anticonvulsant and mood-stabilizing drug valproic acid was identified as an inhibitor of HDAC1, thereby linking its antiepileptic effects to changes in histone acetylation. As described below, more recent work has revealed that inhibitors of class 1 and 2 HDACs represent novel therapeutic approaches to treat neurodegenerative disorders, depression and anxiety, and the cognitive deficits that go hand in hand with many neurodevelopmental disorders.
Deacetylation of histone tails leads to the condensation of chromatin and suppression of transcription, whereas acetylation weakens the binding of histone to DNA, facilitating the accessibility of transcription factors and their complexing with RNA polymerase II. The extent of histone acetylation is a dynamic process that is influenced by the antagonistic enzyme activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). The dynamic nature of acetylation of histone proteins is reflected in the short turnover time for acetyl groups in the core histones [40]. Histone acetylation also influences the interaction of nucleosomes with each other, and thus the structure of chromatin itself. In summary, histone acetylation destabilizes chromatin structure and promotes transcription, whereas deacetylation promotes chromatin condensation and the repression of gene expression. The acetyl groups are donated by acetyl coenzyme A in a reaction catalyzed by a family of HATs. Proteins from one of these families of HAT, referred to as “PCAF,” associate with the complex of p300 and cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein binding protein (CBP); the latter complex is an important global transcriptional co-activator in essentially every cell type and is referred to as p300/CBP [40]. In addition to acetylation of histones, another mechanism of transcriptional regulation by PCAF relates to its ability to acetylate nonhistone transcription-related factors. Interestingly, p300/CBP, which stimulates transcription via its interactions with promoter-binding transcription factors, can itself acetylate the amino terminal tails of all the core histone proteins and nonhistone transcription-related proteins [40].
Implications for Treatment in Neurodegenerative Disorders
Alzheimer’s Disease
Alzheimer’s disease (AD) is a slow, progressive dementia that primarily affects the cortex and hippocampus. AD is characterized by the deposit of abnormal filaments of hyperphosphorylated Tau protein in neuronal cytoplasm, known as neurofibrillary tangles, accompanied by extracellular aggregates of large amyloid β protein (Aβ), known as senile plaques [30]. Decreased amounts of methylcytosine within the APP promoter in the DNA from the parietal cortex of individuals without dementia are associated with increasing age [32]. It was also found that the components of the methylation co-repressor complex were markedly decreased in AD brains [31•]. Elevated levels of homocysteine are seen in patients with AD and are associated with deficiency of folate and vitamin B12 [33], which are essential cofactors required to regenerate methionine (the precursor of the methyl donor for DNA methylation) from homocysteine.
Since increased histone acetylation is found in the brains of patients with AD, the use of histone acetyltransferase (HAT) inhibitors may hold some potential for treatment of AD [34]. The use of biomarkers to define an individual’s epigenome may allow drugs to be tailored to specific epigenomes, so that epigenetic switches could be turned back pharmacologically. So far, the use of HDAC inhibitors in humans has not been successful. A recent clinical trial revealed significant worsening of agitation and aggression in AD patients receiving VPA versus placebo [35]. Moreover, the effectiveness of VPA in treating behavioral symptoms in elderly dementia patients with agitation and aggressive behaviors has also been limited; low doses of VPA fail to reduce agitation in all patients, and high doses in some patients lead to unacceptable adverse effects [36, 37]. While HDAC inhibition may have limited benefits in AD patients and dementia, alternative methods of manipulating the general DNA hypomethylation seen in AD may hold greater therapeutic promise.
Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurological disorder characterized by a number of motor and non-motor symptoms [42]. Pathologically, PD is characterized by the loss of dopaminergic neurons in the substantia nigra (SN) pars compacta, and patients frequently have Lewy bodies, eosinophilic intracellular inclusions composed of amyloid-like fibers and α-synuclein (SNCA) [28]. Misfolded α-synuclein bound to ubiquitin forms a complex that cannot be transferred to the proteasome and aggregates in cytoplasmic inclusions, or Lewy bodies. These inclusions may be a cause of the midbrain dopaminergic neuron loss in the SN [43].
Recently, DNA methylation of human SNCA intron 1 and demethylation of the SNCA CpG in the brains of PD patients was found to regulate SNCA gene expression [44•, 45]. Alternatively, or in addition, nuclear α-synuclein decreases histone H3 acetylation through direct interaction with histones [29]. In vitro cell culture and Drosophila studies have shown that familial PD mutations in α-synuclein (A30P and A53T) cause increased nuclear targeting of α-synuclein in cultured cells. α-synuclein is found to bind to histones and HATs and inhibits acetylation in HAT assays [29].
Consistent with these findings, the administration of class I HDAC inhibitors, such as SAHA (Vorinostat) and sodium butyrate, ameliorate α-synuclein-induced neurotoxicity; these inhibitors have neuroprotective properties in neurotoxin models of PD-related neurodegeneration [29, 46, 47]. In terms of target genes that are potentially affected by epigenetic dysregulation in PD, brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is known to play a key role in the growth, survival, synaptic plasticity, and maintenance of neurons. BDNF expression is regulated by histone acetylation as well as DNA methylation [38]. In the substantia nigra pars compacta of PD-affected neurons, the amount of BDNF mRNA expression is decreased. The pathogenic α-synuclein mutations A30P and A53T are suspected of being linked to the loss of BDNF [39, 40], whereas HDAC inhibitors may rescue BDNF expression by increasing BDNF expression. Recently, inhibition of the SIRT2 NAD-dependent lysine deacetylase was shown to protect against α-synuclein-mediated toxicity in vitro in cellular models and in a Drosophila model of PD [41]; however, the mechanism by which inhibition of SIRT2 confers neuroprotection remains poorly understood, although most recent studies suggest it is through a non-nuclear/histone-mediated effect involving regulation of sterol biosynthesis [48••].
Huntington’s Disease
Huntington’s disease (HD) is a late-onset, autosomal dominant neurodegenerative disorder that presents with prominent motor, cognitive, and psychiatric symptomatology. The disease is associated with expansion of polyglutamine (polyQ) repeats in the amino-terminal portion of huntingtin, a predominantly cytosolic protein of more than 38 repeats; polyglutamine repeats under 35 are not linked to manifest disease [48••]. There is mounting evidence to suggest that transcriptional dysregulation appears to play a significant role in the pathophysiology of HD [24, 25], and may correlate with epigenetic dysregulation.
The therapeutic potential of drugs could be used to manipulate the epigenetic dysregulation seen in HD models and patients. A transgenic Drosophila model exhibited marked histone hypoacetylation corresponding with the transcriptional repression resulting from CBP inactivation. Furthermore, HDAC inhibitors prevented polyQ-induced toxicity and neurodegeneration in a Drosophila model of HD [26, 27], pointing to a novel target for HD therapy. HD transgenic models expressing exon 1 of the human huntingtin gene generally exhibit decreased histone acetylation, which correlates with patterns of reduced gene expression. For example, the HDAC inhibitor phenyl butyrate could increase histone H3 and H4 acetylation and improved the survival rate of HD mice in a dose-dependent manner [49]. Sodium butyrate treatment induced hyperacetylation and reduced neural and brain atrophy and improved motor performance [50]. These studies reveal great therapeutic promise for HDAC inhibitors in the treatment of HD. Conversely, the relation between histone modifications and gene expression is complex. Treatment with HDAC inhibitors in transgenic HD mice induced histone hyperacetylation, but decreased the expression of specific genes associated with cell death [51]. One explanation for the paradoxical changes in gene expression induced by butyrate may be due to the upregulation of a repressor, which in turn reduces the expression of the genes it regulates.
In human HD brain studies, Anderson et al. [52] uncovered interesting evidence for the potential role of histone hyperacetylation, in contrast to the hypoacetylation observed in transgenic Drosophila and rodent models. They saw significant increases in HAT 1 and in histone H3 family 3B mRNA expression in HD brain striatum and cortex, respectively. They also showed gene repression in specific gene clusters, such as Chr1p34, Chr17q21, and ChrXp11.2, all of which encode HDAC genes (HDAC 1, 5, and 6, respectively). The results point to possible species differences among transgenic Drosophila models, rodent models, and the human disease state.
Although the various results can be complicated and at times contradictory, there is ample evidence that alterations in epigenetic pathways are involved in the etiology of HD. Most studies, which use either cell lines or transgenic animals expressing polyQ, have found evidence of reduced histone acetylation. HDAC inhibitors generally provide positive outcomes in these models, although thus far their effects in humans with HD have been much less impressive. Some human studies hint at increased histone acetylation; however, this runs counter to the work using cell lines and transgenic animals. It will be vital in future studies to investigate and compare specific histone modifications in the model systems and the human condition to determine which, if any, of the models are valid.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) presents as a familial and sporadic neurodegenerative disorder characterized by progressive muscle wasting that leads to paralysis due to loss of upper and lower motor neurons. About 10 % of patients report a family history, and mutations in SOD1 (which encodes superoxide dismutase-1) are the cause in about 20 % of these cases.
Transgenic rodent models of ALS expressing mutant SOD1 have been created to unravel pathogenic mechanisms and screen and identify potential therapeutic interventions [53]. The transgenic ALS mice show hypoacetylation of H-4 and other histones in spinal cord sections, and treatment of transgenic ALS mice with sodium phenylbutyrate, an HDAC inhibitor, extended their survival and improved body weight and motor performance. Moreover, neuropathologic examination of the lumbar spinal cord showed less gross atrophy and ventral neuron loss, less reactive gliosis, and reduced immunoreactive staining of ubiquitin-positive aggregates in the cytosol.
Two genes have recently been linked to familial forms of ALS: TARDBP (also known as TDP-43; which encodes TAR DNA-binding protein) [54] and FUS (RNA-binding protein fusion) [55], which comprise 3 % and 4 % of familial cases, respectively, and are generally involved in regulating many aspects of RNA metabolism [56]. Sporadic amyotrophic lateral sclerosis, on the other hand, has been linked to another gene, elongator protein 3 (ELP3), which is important for paternal DNA demethylation [57••]. Knockdown of ELP3 impairs paternal DNA demethylation as indicated by reporter binding, immunostaining, and bisulfite sequencing. The demethylation process may be mediated through a reaction that requires an intact radical SAM domain. ELP3 encodes the catalytic subunit of the HAT complex [58] and can regulate the expression of Hsp70 via the modification of histone H3K14 and H4K8 acetylation [59]. Overexpression of Hsp70 helps to clear the aggregates that arise in a SOD1 mouse model [60] and also protects against mutant SOD1-induced death in mammalian neuronal cells [61]. As with most neurodegenerative diseases, amyotrophic lateral sclerosis also features cytoplasmic deposits of misfolded proteins, consisting of SOD1, TARDBP, or FUS aggregates. The best-known pathogenic mechanism in ALS is the toxic action of SOD1, which lies in its tendency to produce aggregates [62]. FUS is able to bind CBP and strongly inhibit its HAT activity [63], and overexpression of FUS induces hypoacetylation of histones H3K9 and H3K14 on the CCND1 promoter.
Conclusion
Epigenetic changes play important roles in the pathogenesis of neurodegenerative disorders, with many experimental studies showing the involvement of epigenetic modulations in these diseases. Research has revealed DNA methylation and histone modification profiles in some of the more common neurodegenerative disorders, and there is rapidly mounting evidence linking epigenetic alterations to their development. Some of the DNA methylations and histone modifications seen might prove useful as early disease biomarkers, while they might also be targeted with epigenetic drugs, such as DNMT inhibitors and HDAC inhibitors, or with new preclinical drugs that target histone demethylases, histone methyltransferases, and sirtuins. Understanding the contribution of epigenetic changes to common neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, will give us better molecular tools to improve prognosis, diagnosis, and therapeutic intervention for these disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Wu C, Morris JR. Genes, genetics, and epigenetics: a correspondence. Science. 2001;293(5532):1103–5.
Zaratiegui M, Irvine DV, Martienssen RA. Noncoding RNAs and gene silencing. Cell. 2007;128(4):763–76.
Bai L, Morozov AV. Gene regulation by nucleosome positioning. Trends Genet. 2010;26(11):476–83.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68.
•• Narayan, M. Dragunow. Pharmacology of epigenetics in brain disorders. Br J Pharmacol, 2010. 159(2):285–303. This paper reported the pharmacology of epigenetics, such as DNA methyl transferase inhibitors and histone deacetylase inhibitors, to treat brain disorders of the epigenome whether induced developmentally or manifested/acquired later in life.
Esteller, M., Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet, 2007. 16 Spec No 1:R50–9.
• Moore LD, Le T, Fan G. DNA Methylation and Its Basic Function. Neuropsychopharmacology, 2012[Epub ahead of print]. This paper described the DNA methylation and demethylation machinery in the nervous system and its association with other epigenetic mechanisms such as histone modifications and noncoding RNAs.
Chuang LS, et al. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277(5334):1996–2000.
Bostick M, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317(5845):1760–4.
Fatemi M, Wade PA. MBD family proteins: reading the epigenetic code. J Cell Sci. 2006;119(Pt 15):3033–7.
Lopez-Serra L, Esteller M. Proteins that bind methylated DNA and human cancer: reading the wrong words. Br J Cancer. 2008;98(12):1881–5.
Rai K, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135(7):1201–12.
Bhutani N, et al. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463(7284):1042–7.
Popp C, et al. Genome–wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–5.
•• Tahiliani M, et al. Conversion of 5–methylcytosine to 5–hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5. This paper showed that TET1 is a 2-oxoglutarate (2OG)- and Fe(II)-dependent enzyme that catalyzes conversion of 5mC to 5-hydroxymethylcytosine (hmC) in cultured cells and in vitro.
Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–33.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–30.
Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–3.
• Guo JU, et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–34. This paper reported that TET1 promotes DNA demethylation in mammalian cells through a process that requires the base excision repair pathway. DNA demethylation of both exogenously introduced and endogenous 5hmCs is promoted by the AID/APOBEC family of cytidine deaminases. Furthermore, Tet1 and Apobec1 are involved in neuronal activity-induced, region-specific, active DNA demethylation and subsequent gene expression in the dentate gyrus of the adult mouse brain in vivo.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Leng C, et al. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006;34(6):776–87.
Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–81.
Agger K, et al. The emerging functions of histone demethylases. Curr Opin Genet Dev. 2008;18(2):159–68.
Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19(5):233–8.
Hodges A, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15(6):965–77.
Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413(6857):739–43.
Pallos J, et al. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2008;17(23):3767–75.
Weintraub D, Comella CL, Horn S. Parkinson’s disease—Part 1: Pathophysiology, symptoms, burden, diagnosis, and assessment. Am J Manag Care. 2008;14(2 Suppl):S40–8.
Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15(20):3012–23.
Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403.
• Mastroeni D, et al. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31(12):2025–37. This paper evaluated immunoreactivity for two markers of DNA methylation and eight methylation maintenance factors with highly significant decrements in AD cases in entorhinal cortex layer II. These decrements were particularly marked in PHF1/PS396 immunoreactive, neurofibrillary tangle-bearing neurons.
Tohgi H, et al. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70(2):288–92.
Zhuo JM, Wang H, Pratico D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci. 2011;32(9):562–71.
Narayan PJ, Dragunow M. High content analysis of histone acetylation in human cells and tissues. J Neurosci Methods. 2010;193(1):54–61.
Herrmann N, et al. A placebo-controlled trial of valproate for agitation and aggression in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;23(2):116–9.
Tariot PN, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer Disease complicated by agitation: a randomized, controlled trial. Am J Geriatr Psychiatry. 2005;13(11):942–9.
Forester B, et al. Report on an open-label prospective study of divalproex sodium for the behavioral and psychological symptoms of dementia as monotherapy and in combination with second-generation antipsychotic medication. Am J Geriatr Pharmacother. 2007;5(3):209–17.
Martinowich K, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–3.
Kohno R, et al. BDNF is induced by wild-type alpha-synuclein but not by the two mutants, A30P or A53T, in glioma cell line. Biochem Biophys Res Commun. 2004;318(1):113–8.
Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5(6):311–22.
Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516–9.
Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–76.
Migliore L, Coppede F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res. 2009;667(1–2):82–97.
• Jowaed A, et al. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci. 2010;30(18):6355–9. This paper found that methylation of human SNCA intron 1 decreased gene expression, while inhibition of DNA methylation activated SNCA expression.
Matsumoto L, et al. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One. 2010;5(11):e15522.
Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8(1):57–64.
Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26(28):7502–12.
•• Luthi-Carter R, et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc Natl Acad Sci U S A. 2010;107(17):7927–32. This paper demonstrated that Genetic or pharmacologic inhibition of SIRT2 in a striatal neuron model of HD resulted in significant down-regulation of RNAs responsible for sterol biosynthesis via decreased nuclear trafficking of SREBP-2, and achieved neuroprotection.
Gardian G, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280(1):556–63.
Ferrante RJ, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23(28):9418–27.
Thomas EA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008;105(40):15564–9.
Anderson AN, et al. Chromosomal profiles of gene expression in Huntington’s disease. Brain. 2008;131(Pt 2):381–8.
Ryu H, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93(5):1087–98.
Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Kwiatkowski Jr TJ, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–8.
Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–64.
•• Okada Y, et al. A role for the elongator complex in zygotic paternal genome demethylation. Nature. 2010;463(7280):554–8. This paper developed a live cell imaging system to monitor the paternal DNA methylation state in zygotes. Through short-interfering-RNA-mediated knockdown in mouse zygotes, they identified Elp3, a component of the elongator complex, to be important for paternal DNA demethylation. Importantly, injection of messenger RNA encoding the Elp3 radical SAM domain mutant into MII oocytes before fertilization also impaired paternal DNA demethylation, indicating that the SAM radical domain is involved in the demethylation process.
Simpson CL, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet. 2009;18(3):472–81.
Han Q, et al. Gcn5- and Elp3-induced histone H3 acetylation regulates hsp70 gene transcription in yeast. Biochem J. 2008;409(3):779–88.
Koyama S, et al. Alteration of familial ALS-linked mutant SOD1 solubility with disease progression: its modulation by the proteasome and Hsp70. Biochem Biophys Res Commun. 2006;343(3):719–30.
Patel YJ, et al. Hsp27 and Hsp70 administered in combination have a potent protective effect against FALS-associated SOD1-mutant-induced cell death in mammalian neuronal cells. Brain Res Mol Brain Res. 2005;134(2):256–74.
Pasinelli P, et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43(1):19–30.
Wang X, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454(7200):126–30.
Acknowledgments
P. Jin is supported by a grant from the National Institutes of Health (NIH).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xu, Z., Li, H. & Jin, P. Epigenetics-Based Therapeutics for Neurodegenerative Disorders. Curr Tran Geriatr Gerontol Rep 1, 229–236 (2012). https://doi.org/10.1007/s13670-012-0027-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13670-012-0027-0