
Abstract
Uncarialines A-E (1–5), five undescribed monoterpene indole alkaloids, together with five known analogues were obtained from the stems of Uncaria rhynchophylla. Alkaloids 1–3 were unique 3,4-seco-tricyclic alkaloids with a 6/5/10 ring system, while 4 and 5 possessed a rare rearranged scaffold originated from corynantheine-type alkaloids with C-2/C-7 oxidation. Their structures were characterized by a comprehensive analysis of MS, NMR, and ECD. Their effects on blood clotting times of human plasma were evaluated and alkaloid 5 had a slight prolongation effect on both thrombin time and activated partial thromboplastin time (p < 0.001).
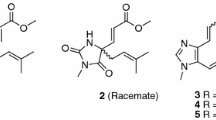
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Monoterpene indole alkaloids (MIAs) are a class of intriguing natural products, which are characterized by structural diversity and promising bioactivities [1,2,3,4,5]. The genus Uncaria (Rubiaceae family) is widely available in tropical regions and there are 14 species in southeast China [1, 6]. The genus Uncaria is enriched with MIAs with anticoagulant [7,8,9], anti-hypertensive [10], anti-Alzheimer’s disease [11], anti-inflammatory [12] and sedative effect [13]. Uncaria rhynchophylla (named “Gou Teng”) was conventionally used for treatment of cardiovascular and cerebrovascular diseases [14]. Interestingly, recent studies have shown that rhynchophylline and isorhynchophylline have anticoagulant effects that delay thrombosis [8, 9]. Besides, many novel MIAs with structural complexity have been characterized from U. rhynchophylla [15,16,17,18,19]. In order to discover structurally novel and biologically active MIAs, uncarialines A-E (1–5), five undescribed MIAs, as well as five known analogues, namely uncarialins D-G (6–9) and dihydrocorynantheine (10) were isolated from the stems of U. rhynchophylla [20, 21]. Presented herein are the isolation, identification, biosynthesis pathway and the anticoagulant activity of uncarialines A-E (1–5).
2 Results and discussion
2.1 Structure elucidation of the compounds
Compound 1 was isolated as a pale-yellow solid. The molecular formula of C23H30N2O4 was established by HRESIMS data (found: m/z 399.2273 [M + H]+, calcd for 399.2278). The observation of IR absorptions at 3423 and 1701 cm−1 implied the existence of amino and ester carbonyl, respectively. NMR spectral data (Tables 1 and 2) demonstrated the existence of an indole moiety (δC 134.6, 112.1, 127.5, 118.9, 119.2, 122.2, 110.9, 136.5), a β-methoxyacrylate methyl ester moiety [δH 7.11 (1H, H-17), 3.61 (3H, 17-OMe), 3.59 (3H, 22-OMe); δC 113.8 (C-16), 158.7 (C-17), 168.5 (C-22), 61.2 (17-OMe), 51.0 (22-OMe)], and a vinyl group [δH 4.82 (1H, H-18b), 4.89 (1H, H-18a), 5.49 (1H, H-19), 2.76 (1H, H-20); δC 114.2 (C-18), 140.2 (C-19), 46.9 (C-20)]. The key 1H-1H COSY cross-peaks of H-3/H-14, H-14/H-15, H-15/H-20, H-20/H-21, H-5/H-6 and HMBC correlations of H-21 (δH 2.59, 2.87) to C-5 (δC 45.8), H-6 (δH 2.94, 3.10) to C-2 (δC 134.6), H-3 (δH 4.79) to C-7 (δC 112.1) indicated the existence of indole-azecane fused heterocycles. Meanwhile, HMBC correlations from H-17 (δH 7.11) to C-15 (δC 31.2) implied that the β-methoxyacrylate methyl ester moiety was linked to C-15. The assignment of a vinyl moiety attached to C-20 was verified by 1H-1H COSY correlations of H-18/H-19/H-20. HMBC correlation of 3-OMe (δH 3.18) to C-3 (δC 75.0) implied that the methoxy group was attached to C-3. The planar structure of uncarialine A was thereby finally established (Fig. 1). The ROESY correlation of H-3 and H-20 indicated both protons were β-oriented, and thus H-15 took α-orientation due to the steric hindrance of the azecane ring. Moreover, the only ROESY correlations of H-17 with 17-OMe established (E)-configuration of the Δ16(17) double bond (Fig. 3). The calculated ECD data of (3S, 15S, 20R)-1 was compatible with its experimental data indicating the correct assignment of the absolute configuration of uncarialine A (1) (Fig. 4, Supplementary file).

Molecular structures of compounds 1–10
Compound 2 was isolated as a pale-yellow solid. It had a molecular formula of C23H32N2O4 in terms of HRESIMS ion at m/z 401.2441 ([M + H]+, calcd for 401.2435). The NMR data of 2 indicated that 2 had the same basic scaffold as that of 1 (Tables 1 and 2). The distinct observations were the presence of an ethyl group (δH 0.86, δC 12.4; δH 0.85, 1.35, δC 21.9) in 2. The molecular weight of 2 has two more mass units and one less unsaturation than that of 1, demonstrating 2 was the Δ18(19) double bond reduction form of 1. The structure of uncarialine B was thereby established (Fig. 1), which was further verified by HMBC and 1H-1H COSY spectra analysis (Fig. 2). The identical ROESY and ECD spectra of uncarialine B (2) and alkaloid 1 demonstrated both alkaloids had the same relative and absolute configurations (Figs. 3 and 4).

Key HMBC (arrow) and 1H-1H COSY (bold) correlations of uncarialines A-E (1–5)

Key ROESY correlations of uncarialines A-E (1–5)

Experimental and calculated ECD of uncarialines A-E (1–5)
Compound 3 was isolated as a pale-yellow solid. It had the molecular formula of C24H32N2O4 by HRESIMS analysis (found: m/z 413.2443 [M + H]+, calcd for 413.2435) with 14 mass units larger than that of 1. The NMR data of 3 indicated that 3 had the same basic scaffold as that of 1 (Tables 1 and 2), with the existence of a unique signal with N-methyl [δH 2.16 (3H, s, N-Me); δC 40.5 (N-Me)], and an allyl group [δH 1.76 (3H, H-18), 5.47 (1H, H-19); δC 13.5 (C-18), 127.9 (C-19), 138.1 (C-20)]. HMBC correlation of N-Me (δH 2.16) with C-5 (δC 55.0) and C-21 (δC 65.0) indicated that the methyl group was linked to N-4. Moreover, the allyl attached to C-20 due to the HMBC correlations of H-18 (δH 1.76) to C-20 (δC 138.1). Thus, the structure of uncarialine C was thereby established (Fig. 1). The ROESY cross-peaks of H-18 with H-21b and of H-17 with 17-OMe, confirmed (Z)- and (E)-configurations of Δ19(20) and Δ16(17) double bonds, respectively. The deficiency of ROESY correlation of H-15 with H-3 confirmed that the methoxy group (C-3) and the β-methoxyacrylate methyl ester moiety (C-15) were opposite. Thus, there are two possible stereoisomers (3R*,15R*)-3 or (3S*,15S*)-3. The absolute stereochemistry of (3R,15R)-3 was assigned finally by the compatible calculated and experimental ECD spectra of uncarialine C (3) (Fig. 4).
Compound 4 was isolated as a white solid. It had the molecular formula of C22H28N2O5, as evidenced by HRESIMS ion at m/z 401.2076 ([M + H]+, calcd for 401.2071). The maxima UV absorptions at 207, 242, and 295 nm demonstrated an oxindole chromophore [22]. IR absorptions showed the existence of amide carbonyl (1625 cm−1), ester carbonyl (1708 cm−1), and amino group (3423 cm−1). 13C NMR spectroscopy suggested that 4 had 22 carbons and had a high similarity with uncarialin D [20], except for the terminal vinyl group in uncarialin D was reduced to ethyl group in 4. Meanwhile, the ROESY correlations from H-15 to H-3 and H-19b indicated H-3 and H-15 were α-oriented while H-20 was β-oriented. Moreover, the only ROESY correlation of H-17 with 17-OMe in ROESY spectrum indicated the Δ16(17) double bond took (E)-configuration. The Cotton effects at 212, 269, and 248 nm suggested the absolute configuration of (3R,7R,15S,20R)-4 [23, 24], which was confirmed by the ECD calculation of uncarialine D (4) (Fig. 4).
Compound 5 was isolated as a white solid. It had a molecular formula of C21H24N2O5, as given by HRESIMS analysis (found: m/z 385.1758 [M + H]+; calcd for 385.1758). The IR absorptions implied the existence of amino group (3422 cm−1), ester carbonyl (1709 cm−1) and amide carbonyl (1626 cm−1). Interpretation of its NMR data suggested 5 had a similarity with melodinoxanine [25]. The major difference was that 5 lacked two aromatic methoxy groups, which was further verified by the key 1H-1H COSY cross-peaks of H-9/H-10, H-10/H-11, and H-11/H-12. The ROESY correlations of H-3 with H-15, and H-21a confirmed that they were assigned as α-oriented. Thus, the ROESY cross-peaks of H-19 with H-14 and H-21b implied H-19 took β-orientation. The coupling constant (J19,20 = 12.5 Hz) between H-19 and H-20 in the 1H NMR spectrum confirmed H-20 took α-orientation. Additionally, the only ROESY cross-peaks of H-17 with 17-OMe indicated (E)-configuration of the Δ16(17) double bond. The absolute stereochemistry of uncarialine E (5) was finally characterized by the ECD calculation result of (3R,7R,15S,19S,20S)-5 identical with the corresponding experimental ECD data (Fig. 4).
The possible biogenetic routes for 1–5 is presented in Scheme 1. Biogenetically, the H-3 of uncarialin A or dihydrocorynantheine (10) was initially oxidized to hydroxyl group, and then the hydroxyl derivatives undergo hydrolysis of tertiary amine under acidic conditions to yield intermediates i and ii, which eventually undergoes reduction, oxidation, and methylation to form compounds 1–3, respectively. Likewise, dihydrocorynantheine (10) was oxidized to 2,7-dihydroxy-dihydrocorynantheine N-oxide, followed by hydrolysis of quaternary ammonium under acidic conditions to form the key intermediate iii, which finally undergoes rearrangement reaction to form compounds 4 and 5.

Hypothesis biogenetic pathway for uncarialines A-E (1–5)
2.2 Anticoagulant activity
The anticoagulant activity of the new isolates is represented by the following parameters: thrombin time (TT), prothrombin time (PT) and activated partial thromboplastin time (APTT) [26]. Compounds 1–4 were inactive on TT, PT and APTT (p > 0.05), while compound 5 had a slightly prolongation effect on both TT and APTT (p < 0.001) (Table 3).
3 Experimental
3.1 General experimental procedures
The experimental apparatus is as previously reported [2, 3]
3.2 Plant material
The stems of U. rhynchophylla were obtained in Jianhe, Guizhou Province, China, on May 2020 and identified by Prof. Hongping He, one of our co-authors. The sample specimen (No. Z20200520) was deposited at Kunming Institute of Botany.
3.3 Extraction and isolation
The crushed stems of U. rhynchophylla were cold soaked 3 times in methanol (MeOH) to obtain the extract. The crude alkaloids (2030 g) were obtained by the previously procedures [2, 3], which were divided to six fractions (A-F) using silica gel column chromatography (DCM/MeOH, 49:1, 29:1, 9:1, 1:1, v/v). Among them, fraction B (18 g) was divided into three fractions (B1–B3) by a silica gel column (300–400 mesh, DCM/MeOH, 49:1, 29:1, 9:1, 1:1, v/v). Fraction B2 (1.8 g) was separated by HPLC with MeCN/H2O (60:40, 0.01% Et2NH, 3 mL/min) to give 5 (13 mg, tR 15.0 min) and 10 (47 mg, tR 21.0 min). Fraction C (96 g) was divided to seven fractions (C1-C7) by a silica gel column (DCM/MeOH, 49:1, 19:1, 9:1, 1:1, v/v). Fraction C3 (4.2 g) was separated by RP-C18 (MeOH/H2O, 30:70, 50:50, 100:0, v/v) and HPLC with MeCN/H2O (52:48, 0.01% Et2NH, 3 mL/min) to give 4 (5 mg, tR 11.0 min) and 6 (7 mg, tR 28.0 min). Fraction E (184 g) was divided to nine fractions (E1-E9) by silica gel column chromatography (DCM/MeOH, 19:1, 9:1, 1:1, v/v). Fraction E2 (520 mg) was separated by Sephadex LH-20 (MeOH) and HPLC with MeCN/H2O (45:55, 0.01% Et2NH, 3 mL/min) to obtain 1 (9 mg, tR 9.0 min) and 2 (11 mg, tR 18.0 min). Fraction E3 (210 mg) was further separated by HPLC with MeCN/H2O (36:64, 0.01% Et2NH, 3 mL/min) to give 7 (26 mg, tR 13.0 min), 8 (34 mg, tR 20.5 min) and 9 (33 mg, tR 27.0 min). Fraction E5 (2.5 g) was separated by Sephadex LH-20 and subsequent HPLC separation with MeCN/H2O (30:70, 0.01% Et2NH, 3 mL/min) to obtain 3 (43 mg, tR 38.0 min).
3.4 Uncarialine A (1)
Uncarialine A (1): pale-yellow solid; \({[\alpha]^{22}_{\text{D}}}\) − 8 (c 0.3, MeOH); UV (MeOH) λmax (log ε): 223 (3.4) nm; ECD (0.0034 M, MeOH) λmax (∆ε) 230 (+ 11.9), 271 (− 1.9); IR (KBr) vmax 3423, 2922, 2852, 1701, 1634, 1461, 1244, 1116 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Tables 1 and 2; HRESIMS m/z 399.2273 [M + H]+ (calcd for C23H31N2O4, 399.2278).
3.5 Uncarialine B (2)
Uncarialine B (2): pale-yellow solid; \({[\alpha]^{22}_{\text{D}}}\) − 7 (c 0.3, MeOH); UV (MeOH) λmax (log ε): 223 (3.3) nm; ECD (0.0048 M, MeOH) λmax (∆ε) 230 (+ 11.4), 271 (− 2.0); IR (KBr) vmax 3429, 2922, 2852, 1701, 1632, 1461, 1244, 1106 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Tables 1 and 2; HRESIMS m/z 401.2441 [M + H]+ (calcd for C23H33N2O4, 401.2435).
3.6 Uncarialine C (3)
Uncarialine C (3): pale-yellow solid; \({[\alpha]^{22}_{\text{D}}}\)+ 184 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 228 (3.6) nm; ECD (0.0023 M, MeOH) λmax (∆ε) 199 (+ 17.6), 230 (− 6.7), 247 (+ 3.8), 287 (+ 2.6); IR (KBr) vmax 3420, 2938, 1705, 1634, 1461, 1236, 1093 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Tables 1 and 2; HRESIMS m/z 413.2443 [M + H]+ (calcd for C24H33N2O4, 413.2435).
3.7 Uncarialine D (4)
Uncarialine D (4): white solid; \({[\alpha]^{22}_{\text{D}}}\) + 7 (c 0.2, MeOH); UV (MeOH) λmax (log ε): 207 (3.6) nm; ECD (0.0021 M, MeOH) λmax (∆ε) 212 (+ 30.8), 248 (− 11.4), 269 (+ 7.4); IR (KBr) vmax 3423, 2924, 2853, 1708, 1625, 1470, 1247, 1108 cm−1; 1H and 13C NMR data (CD3OD, 600 and 150 MHz) see Tables 1 and 2; HRESIMS m/z 401.2076 [M + H]+ (calcd for C22H29N2O5, 401.2071).
3.8 Uncarialine E (5)
Uncarialine E (5): white solid; \({[\alpha]^{22}_{\text{D}}}\) − 32 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 208 (4.3) nm; ECD (0.0003 M, MeOH) λmax (∆ε) 210 (+ 32.8), 241 (− 19.6), 270 (+ 4.8); IR (KBr) vmax 3422, 2927, 2854, 1709, 1626, 1472, 1211, 1100 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Tables 1 and 2; HRESIMS m/z 385.1758 [M + H]+ (calcd for C21H25N2O5, 385.1758).
3.9 Blood clotting times
Chemicals. Reagents of TT, PT and APTT, CaCl2 and coagulation control plasma were produced in TECO (Germany). Tris–HCl was purchased from Amresco (USA). Reference anticoagulant drug (heparin, HEP; low molecular weight heparin, LMWH) and DMSO were produced in Sigma-Aldrich (USA).
The measurements were taken using the MC-4000 Optic coagulometer (Germany). Prior to the detection, coagulation control plasma were pre-incubated with the examined compounds (15 min, 37 °C) at the final concentrations of 200 μM.
4 Concluding remarks
In summary, 10 alkaloids including five new ones were obtained from the stems of U. rhynchophylla. Among them, uncarialines A-C (1–3) were unique 3,4-seco-tricyclic MIAs with a 6/5/10 ring system, while uncarialines D (4) and E (5) possessed a rare rearranged skeleton derived from corynantheine-type alkaloids with C-2/C-7 oxidation. It is noteworthy that the stereochemistry of 3 at C-3 and C-15 were opposite to those of 1 and 2 indicating the possibility of specific enzyme catalyze the formation of the corresponding chiral centers. The findings not only enrich the diversity of secondary metabolisms of U. rhynchophylla, but only provide insight into the complex biosynthetic mechanism of such alkaloids category.
Change history
12 May 2023
A Correction to this paper has been published: https://doi.org/10.1007/s13659-023-00378-z
References
Ndagijimana A, Wang XM, Pan GX, Zhang F, Feng H, Olaleye O. A review on indole alkaloids isolated from Uncaria rhynchophylla and their pharmacological studies. Fitoterapia. 2013;86:35–47.
Zhang Y, Bai X, Yuwen HS, Guo LL, Liu JW, Hao XJ. Alkaloids from Tabernaemontana divaricata combined with fluconazole to overcome fluconazole resistance in Candida albicans. Bioorg Chem. 2021;107: 104515.
Zhang Y, Yuan YX, Goto M, Guo LL, Li XN, Morris-Natschke SL, Lee KH, Hao XJ. Taburnaemines A-I, cytotoxic vobasinyl-iboga-type bisindole alkaloids from Tabernaemontana corymbosa. J Nat Prod. 2018;81:562–71.
Faisal S, Badshah SL, Kubra B, Emwas AH, Jaremko M. Alkaloids as potential antivirals. A comprehensive review. Nat Prod Bioprospect. 2023;13:4.
Sun SF, Zhong HJ, Zhao YL, Ma XY, Luo JB, Zhu L, Zhang YT, Wang WX, Luo XD, Geng JW. Indole alkaloids of Alstonia scholaris (L.) R. Br. alleviated nonalcoholic fatty liver disease in mice fed with high-fat diet. Nat Prod Bioprospect. 2022;12:14.
Zhang Q, Zhao JJ, Xu J, Feng F, Qu W. Medicinal uses, phytochemistry and pharmacology of the genus Uncaria. J Ethnopharmacol. 2015;173:48–80.
Horie S, Yano S, Aimi N, Sakai S, Watanabe K. Effects of hirsutine, an antihypertensive indole alkaloid from Uncaria rhynchophylla, on intracellular calcium in rat thoracic aorta. Life Sci. 1992;50:491–8.
Zou L, Lu FY, Lin B, Zhou Y, Liu TT, Sun Y. Stability of alkaloids during drying process and their effect on anticoagulating activity of Uncariae Ramulus Cum Uncis. J Anal Methods Chem. 2019;2019:7895152.
Sakti AS, Nityasa AR, Saputri FC. Effect of Uncaria gambir and Uncaria sclerophylla on pulmonary-thromboembolism mice. Phcog J. 2020;12:192–6.
Gao L, Kong X, Wu W, Feng Z, Zhi H, Zhang Z, Long H, Lei M, Hou J, Wu W. Dissecting the regulation of arachidonic acid metabolites by Uncaria rhynchophylla (Miq). Miq. in spontaneously hypertensive rats and the predictive target sEH in the anti-hypertensive effect based on metabolomics and molecular docking. Front Pharmacol. 2022;13:909631.
Lu JH, Tan JQ, Durairajan SSK, Liu LF, Zhang ZH, Ma L, Shen HM, Chan HE, Li M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy. 2012;8:98–108.
Azevedo BC, Morel LJF, Carmona F, Cunha TM, Contini SHT, Delprete PG, Ramalho FS, Crevelin E, Bertoni BW, França SC. Aqueous extracts from Uncaria tomentosa (Willd. ex Schult.) DC. reduce bronchial hyperresponsiveness and inflammation in a murine model of asthma. J Ethnopharmacol. 2018;218:76–89.
Sakakibara I, Terabayashi S, Kubo M, Higuchi M, Komatsu Y, Okada M, Taki K, Kamei J. Effect on locomotion of indole alkaloids from the hooks of Uncaria plants. Phytomedicine. 1999;6:163–8.
Qin N, Lu X, Liu Y, Qiao Y, Qu W, Feng F, Sun H. Recent research progress of Uncaria spp. based on alkaloids: phytochemistry, pharmacology and structural chemistry. Eur J Med Chem. 2021;210:112960.
Zhou HF, Li WY, Peng LY, Li XN, Zuo ZL, Zhao QS. Rhynchines A-E: Cav3.1 calcium channel blockers from Uncaria rhynchophylla. Org Lett. 2021;23:9463–7.
Xu Y, Wang R, Hou T, Li H, Han Y, Li Y, Xu L, Lu S, Liu L, Cheng J. Uncariphyllin A-J, indole alkaloids from Uncaria rhynchophylla as antagonists of dopamine D2 and Mu opioid receptors. Bioorg Chem. 2023;130: 106257.
Yu ZL, Bai R, Zhou JJ, Huang HL, Zhao WY, Huo XK, Yang YH, Luan ZL, Zhang BJ, Sun CP. Uncarialins J-M from Uncaria rhynchophylla and their anti-depression mechanism in unpredictable chronic mild stress-induced mice via activating 5-HT1A receptor. Chin J Chem. 2021;39:1331–43.
Guo Q, Si X, Shi Y, Yang H, Liu X, Liang H, Tu P, Zhang Q. Glucoconjugated monoterpene indole alkaloids from Uncaria rhynchophylla. J Nat Prod. 2019;82:3288–301.
Guo Q, Yang H, Liu X, Si X, Liang H, Tu P, Zhang Q. New zwitterionic monoterpene indole alkaloids from Uncaria rhynchophylla. Fitoterapia. 2018;127:47–55.
Liang JH, Luan ZL, Tian XG, Zhao WY, Wang YL, Sun CP, Huo XK, Deng S, Zhang BJ, Zhang ZJ. Uncarialins A-I, monoterpenoid indole alkaloids from Uncaria rhynchophylla as natural agonists of the 5-HT1A receptor. J Nat Prod. 2019;82:3302–10.
Wenkert E, Cochran D, Hagaman E, Schell F, Neuss N, Katner A, Potier P, Kan C, Plat M, Koch M. Carbon-13 nuclear magnetic resonance spectroscopy of naturally occurring substances. XIX. Aspidosperma alkaloids. J Am Chem Soc. 1973;95:4990–5.
Lim KH, Sim KM, Tan GH, Kam TS. Four tetracyclic oxindole alkaloids and a taberpsychine derivative from a Malayan Tabernaemontana. Phytochemistry. 2009;70:1182–6.
Cao XF, Wang JS, Wang XB, Luo J, Wang HY, Kong LY. Monoterpene indole alkaloids from the stem bark of Mitragyna diversifolia and their acetylcholine esterase inhibitory effects. Phytochemistry. 2013;96:389–96.
Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, Sakai SI. A new indole alkaloid, 7α-hydroxy-7H-mitragynine, from Mitragyna speciosa in thailand. Planta Med. 1994;60:580–1.
Kitajima M, Ohara S, Kogure N. New indole alkaloids from Melodinus henryi. Heterocycles. 2012;85:1949–59.
Kolodziejczyk-Czepas J, Ponczek M, Sady-Janczak M, Pilarski R, Bukowska B. Extracts from Uncaria tomentosa as antiplatelet agents and thrombin inhibitors–the in vitro and in silico study. J Ethnopharmacol. 2021;267: 113494.
Acknowledgements
This work was financially supported by Yunnan Applied Basic Research Projects (No. 202301AS070057), National Key R&D Program of China (No. 2022YFF1100301), and Yunnan Revitalization Talents Support Plan-Young Talent Project (to Y. Zhang).
Author information
Authors and Affiliations
Contributions
K-PH carried out the isolation and the writing of original draft at leading degree. L-LX contributed to isolation at supporting degree. SL contributed to data analysis at supporting degree. Y-LW contributed to investigation, and validation at supporting degree. LY contributed to biological investigation. X-JH contributed to project administration at supporting degree. H-PH contributed to guiding of the writing and data proof reading. YZ contributed to funding acquisition and project administration at leading degree. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: The corresponding authorship was missed for Hong-Ping He, and has been updated correctly.
Supplementary Information
Additional file 1.
HRESIMS, NMR, ECD, and IR spectra of compounds 1–5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, KP., Xu, LL., Li, S. et al. Uncarialines A-E, new alkaloids from Uncaria rhynchophylla and their anticoagulant activity. Nat. Prod. Bioprospect. 13, 13 (2023). https://doi.org/10.1007/s13659-023-00377-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-023-00377-0