
Abstract
Background
The Gram-negative intracellular bacterium Mycoplasma anatis is a pathogen of respiratory infectious diseases in ducks and has caused significant economic losses in the poultry industry.
Objective
This study, as the first report of the structure and function of the pan-genome of Mycoplasma anatis, may provide a valuable genetic basis for many aspects of future research on the pathogens of waterfowl.
Methods
We sequenced the whole genomes of 15 Mycoplasma anatis isolated from ducks in China. Draft genome sequencing was carried out and whole-genome sequencing was performed by the sequencers of the PacBio Sequel and an IonTorrent Personal Genome Machine (PGM). Then the common genic elements of protein-coding genes, tRNAs, and rRNAs of Mycoplasma anatis genomes were predicted by using the pipeline Prokka v1.13.7. To investigate homologous protein clusters across Mycoplasma anatis genomes, we adopted Roary v3.13.0 to cluster orthologous genes (OGs) based on the following criteria.
Results
We obtained one complete genome and 14 genome sketches. Microbial mobile genetic element analysis revealed the distribution of insertion sequences (IS30, IS3, and IS1634), prophage regions, and CRISPR arrays in the genome of Mycoplasma anatis. Comparative genomic analysis decoded the genetic components and functional classification of the pan-genome of Mycoplasma anatis that comprised 646 core genes, 231 dispensable genes and among them 110 was strain-specific. Virulence-related gene profiles of Mycoplasma anatis were systematically identified, and the products of these genes included bacterial ABC transporter systems, iron transport proteins, toxins, and secretion systems.
Conclusion
A complete virulence-related gene profile of Mycoplasma anatis has been identified, most of the genes are highly conserved in all strains. Sequencing results are relevant to the molecular mechanisms of drug resistance, adaptive evolution of pathogens, population structure, and vaccine development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mycoplasma anatis, a Gram-negative bacterium belonging to the family Mycoplasmatacceae under the class Mollicutes, is an intracellular bacterial pathogen of ducks. M. anatis infection can cause serious respiratory diseases, and the main clinical symptoms are sinusitis, air sacculitis, peritonitis, and embryo lethality (Stipkovits and Kempf 1997). The bacteria may also be involved in cloaca and phallus inflammation. Egg infertility is the most common symptom caused by this agent in waterfowl (Stipkovits et al. 1986). Mycoplasma strains can be transmitted vertically (Bencina 1988). M. anatis infection can cause reduced hatching success and growth rates (Bencina 1988) and thus has a serious impact on the economic benefits of waterfowl breeding.
Some gene products related to the pathogenesis of Mycoplasma and immune escape have been described, including adhesin protein, surface lipoproteins, and V-1antigens (Razin 1998). The wall-less mycoplasmas can invade host cells and then replicate and survive as intracellular parasites. Mycoplasma is the smallest self-replicating species known at present. Mycoplasma is a frequent contaminant in mammalian cell culture, and it is also a major problem in the biopharmaceutical industry. In any case, the genetic components involved in the biosynthesis of potential virulence factors of M. anatis have yet to be characterized.
Like other mycoplasmas (Waites et al. 2017), M. anatis is also a “culturally difficult” bacterial species. The rapid diagnosis of duck mycoplasma based on molecular biology requires more specific genetic markers. In recent years, the maturity of NGS technology has greatly improved the speed of obtaining whole-genome sequences of bacteria and has accelerated progress in microbial genetics and ecology. The first complete genome sequence of M. anatis type strain NCTC 10,156 isolated from a diseased duck with sinusitis in 1964 has been recently reported (Dénes et al. 2018). To better characterize the genetic diversity of M. anatis, we sequenced the whole genome and analyzed the comparative genomes of 15 strains isolated from China. The pangenome structure and function of this waterfowl pathogen were comprehensively characterized.
Materials and methods
M. anatis strains
The samples were inoculated in Mycoplasma Broth Base liquid medium (BD Biosciences, San Jose, CA, USA) containing 10% sterile porcine serum, cultured in 5% CO2 at 37 °C in a constant temperature incubator for 8–12 h, filtered with a 0.22 μm filter membrane, adjusted pH to 7.4, supplemented with 2–5 mL FM-4 medium, and cultured in 5% CO2 at 37 °C in a constant temperature incubator until the medium color turned yellow. The designed primer MA was used to identify the strains by PCR, and the primers were as follows: forward primer, 5’-CACGTACTTAACATACCCTTT-3’; reverse primer, 5’-TTCATAACTCTAGTTCGCCAG-3’. Total genomic DNA was extracted using the TIANamp Bacteria DNA kit (TIANGEN).
Genome sequencing, assembly, and annotation
Draft genome sequencing was carried out using the Illumina NovoSeq platform (Illumina, CA). Approximately 1 µg DNA was sheared to construct a sequencing library of ~ 350-bp insertion fragments according to the instructions of the TruSeq™ DNA Sample Prep Kit (Illumina). Paired-end 150-bp reads were then sequenced, producing an average of ~ 8.3 million reads per strain. The sequencing adaptors and low-quality reads were trimmed and filtered using Trimmomatic v0.39 (Bolger et al. 014) with the options ILLUMINACLIP:adapters.fa:2:30:10:5 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 MINLEN:40 AVGQUAL:20. De novo genome assembly was carried out using SPAdes v3.13.1 (Bankevich et al. 2012) with default options. The contigs of more than 500 bp were retained for the subsequent analyses.
Whole-genome sequencing for a selected strain was performed by the sequencers of the PacBio Sequel (Pacific Biosciences, CA) and an IonTorrent Personal Genome Machine (PGM) (Life Technologies, CA). After quality control, the former generated 150,763 long-reads with a mean length of 7 kb, and the latter produced ~ 3.9 million short reads. The integrative pipeline Unicycler v0.4.8 (Bankevich et al. 2012) that consists of state-of-the-art computational tools for read correction, assembly, and polishing was applied to conduct a hybrid assembly of long- and short-reads.
Completeness and contamination of the genome assemblies generated above were assessed by using CheckM v1.0.12 with the options taxonomy_wf genus Mycoplasma (Parks et al. 2015). The genome coverage analysis was performed by using BBmap v38.73 with default options (Bushnell 2014). The newly sequenced genome together with publicly available genomes of M. anatis were annotated using the pipeline Prokka v1.13.7 (Seemann 2014). Briefly, the common genic elements of protein-coding genes, tRNAs, and rRNAs were predicted. The genetic code (–gcode 4) specific to Mycoplasma species was set. Protein functional annotation was supplied by the Swiss-Prot database included in Prokka using BLASTp 2.9.0 + with the option –e value 1e−10 (Altschul et al., 1997). Additionally, the prophage sequence was predicted using the web server PHAST (Zhou et al. 2011). The elements of insertion sequences were detected using ISEScan v1.7.2 with default parameters (Xie 2017). The CRISPR arrays were predicted using MinCED v0.4.0 (Bland et al. 2007) with options -spacers -minNR 4 -minSL 28 -maxSL 53. The sequence similarity analysis for the repeat and spacer elements of CRISPR arrays was conducted using CRISPRCasdb (Pourcel et al., 2020).
Pangenome analysis
To investigate homologous protein clusters across M. anatis genomes, we adopted Roary v3.13.0 (Page et al. 2015) to cluster orthologous genes (OGs) based on the following criteria: minimum percentage identity of 90% with BLASTp; Mycoplasma genetic code; no splitting of paralogs. Three types of genes were summarized: core genes present in all strains, genes present in at least two strains, and strain-specific genes. The binary matrix with the presence and absence of each gene across all strains was used to estimate the sizes of the pan-genome and core-genome by the program PanGP v1.0.1 with the totally random sampling algorithm (Page et al. 2015). Functional classification of the resulting OGs was performed based on the cluster of orthologous groups (COG) database updated in 2014 (Tatusov et al. 2003) and the virulence factor database VFDB updated in 2019 (Tatusov et al. 2003) using BLASTp 2.9.0 + (Altschul et al. 1997). Protein functional domains were searched against the profiles of hidden Markov models (HMMs) curated by the Pfam database v32 using HMMER (Altschul et al. 1997) with the E value cutoff set to 1e−4. Prediction of protein subcellular localization was performed using PSORTb v3.0.2 (Yu et al. 2010) with the option for Mycoplasma species: Gram-negative without outer membrane.
Phylogenetic analyses
For the newly sequenced genomes, species identification was initially performed by calculating average nucleotide identity (ANI) values between the query and closely related representative genomes by PyANI v0.3.0 (Yu et al. 2010) with the option -m ANIb. The genomes were assigned to the representative species according the suggested cutoff of 95% ANI (Richter and Rosselló-Móra 2009). Based on the multiple sequence alignment of core genes created by MAFFT v7.407 (Richter and Rosselló-Móra 2009), a maximum likelihood phylogenetic tree was reconstructed using IQ-TREE (Nguyen et al. 2015) with the inferred best-fitting model of nucleotide substitution and the bootstrap test of 1,000 replicates.
Statistical analyses
Fisher’s exact test was used to analyze the differences between the three gene sets (core, dispensable genes, and strain-specific genes) in the gene count data of each COG functional class. The obtained p-values were corrected for multiple tests using the Bonferroni method, and the significance level of false discovery rate (FDR) was set to 5%.
Results and discussion
General features of 15 M. anatis genomes
Fourteen M. anatis strains were selected for Illumina sequencing to obtain genome sketches. After sequence splicing, the sizes of these genome sketches were about 932–993 kb, with the average GC percentage ranging from 26.7 to 27.0% (Table 1). For these 14 strains of M. anatis, the number of contigs in each genome ranged from 25 to 95. The average N50 of the genome assemblies was 58 kb, ranging from 22 to 123 kb. In addition, M. anatis strain LC01, a potential candidate vaccine strain with the highest titer (> 1010 CCU/ml) strain among the M. anatis strains we isolated, the complete genome was assembled through hybrid assembly of both PacBio and Ion-torrent sequencing data. The genome of LC01 was composed of a 1,012,697 bp circular chromosome, as shown in Fig. 1. The GC% content of the LC01 genome was 26.8%, which was consistent with the full genome of M. anatis type strain NCTC 10,156 (Table 1). The estimates of completeness and contamination of all newly sequenced M. anatis genomes are summarized in Table S1. Based on the gene prediction, the average number of protein coding genes in each genome was 735; the LC01 genome encoded the most CDSs (788), and the DP15 encoded the least number of CDSs (722). With regard to the 15 mycoplasmas isolated from ducks, the general characteristics of genome splicing and their Genbank entry numbers are shown in Table 1.

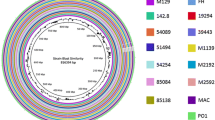
Circular representation of genomic sequence alignments of 16 M. anatis strains. The innermost two rings denote GC content and GC skew plot. The outermost 16 rings display pairwise nucleotide sequence alignments between the complete genome of LC01 as a reference and the genome assemblies of other M. anatis strains. The rings for the genomes are color-coded based on the BLASTn scores (minimum sequence identity of 80% and E value threshold of 1e−10) between the query and reference genomes. The outermost arcs denote the predicted prophage and IS elements as well as the putative genome islands of the LC01 genome
To provide a glimpse of conserved genome regions of M. anatis, Fig. 1 displays a circular sequence alignment map of sequence similarity comparisons between the complete genome of LC01 and the genome assemblies of other M. anatis strains. Pairwise genome alignment showed that the proportion of highly conserved sequences (> 95% identity) compared with the LC01 genome varied from 99.0% (LH05 vs LC01) to 83.8% (NCTC 10,156 vs LC01). The genomic fragments greater than 300 bp that were present in the LC01 genome but absent in the genome of NCTC 10,156 are summarized in Table S1. Two large genomic islands of LC01 were observed, GI 1 (20.6 kb, the genomic coordinates from 438.2 to 458.8 kb) and GI 2 (72.0 kb, 888.0 to 960.0 kb) (Fig. 1). Most of the protein coding genes (21 of 23) on these two GIs are annotated as hypothetical proteins, except that two genes encode extracellular matrix-binding protein Ebh and chromosome partition protein Smc.
In addition, the elements of insertion sequences (ISs) were investigated in the genomes of M. anatis. Three IS elements (IS30, IS3, and IS1634) were identified (Table S2). It appeared that the IS30 element was the most abundant in each of the 16 M. anatis genomes. Comparison of the complete genomes of LC01 and NCTC 10,156 revealed that 34 copies of IS30 and six copies of IS1634 were detected in the LC01 genome, whereas 14 copies of IS30, two copies of IS1634, and nine copies of IS3 were present in the NCTC 10,156 genome. The positions of all predicted ISs in the LC01 genome are shown in Fig. 1. It is worth noting that IS30 exists in the two GIs regions of the LC01 strain mentioned above. Meanwhile, the genomic regions encompassing the IS elements exhibited remarkable genetic differentiation across strains, perhaps due to induction of horizontal gene transfer events. Both families of IS30 and IS3 have been documented as to their likely roles in enhancing genome plasticity of M. bovis (Nguyen et al. 2015). In addition, IS1634 that bears long and variable-length direct repeats has been characterized in M. mycoides subsp. mycoides, a bovine pathogen (Vilei et al. 1999). In contrast, as illustrated in Fig. 1, an incomplete prophage island (12.0 kb, 648,597 to 660,692 kb) that was highly conserved in all the genomes of M. anatis was identified in the LC01 genome. This was composed of 13 protein-coding sequences, 9 of which encoded phage-related proteins.
Phylogeny of M. anatis
First, we used an ANI-based method to determine the species of the newly sequenced mycoplasma strains. Compared with the reference genome of M. anatis type strain NCTC10156, these new genomes had ANI values of 97.40–97.93% (Table 1). The ANI values of these genomes and other mycoplasma species were less than 74%. According to the recommended ANI threshold of 95% (Vilei et al. 1999) at the species boundary, these isolates in this study belong to M. anatis (Fig. S1). The maximum likelihood phylogenetic tree of duck Mycoplasma was constructed by core gene alignment and is displayed in Fig. 2. Obviously, the 15 strains of Mycoplasma isolated from ducks in China were divided into two closely related phylogenetic clades (I and II) according to the region where they were isolated, and neither were closely related to the strain NCTC10156 isolated from the United Kingdom. At the same time, genomic comparisons based on ANI showed that the ANI values between these newly sequenced genomes varied from 98.18 to 99.99%, indicating that these strains of M. anatis isolated from China were more closely related to each other.

Maximum likelihood phylogeny of M. anatis strains. The tree reconstruction is based on the 676,110-nt alignment of all the core genes present in 16 M. anatis genomes. The tree nodes are color-coded with the bootstrap values. The tree is viewed as a rectangular cladogram
Structure and function of the M. anatis pan-genome
To investigate the distribution of homologous protein clusters in M. anatis, we carried out pan-genomic analysis based on 11,816 CDSs encoded by 16 M. anatis genomes. The total number of lineal homologous gene clusters (orthologous genes, OGs) of M. anatis was 869 (Table S3). Of these lineal homologous genes, 646 were identified as core genes. These were genes (Fig. 3A) present in all strains, including 617 single-copy and 29 multi-copy core genes. Among the strains of M. anatis we studied, the proportion of core genes in each genome varied between 81% for NCTC 10,156 to 89% for DP15. In addition, the pan-genome of M. anatis consisted of 121 dispensable genes (exist in at least two strains) and 102 strain-specific genes (genes that exist only in one strain). We further estimated the M. anatis the pan- and core-genome sizes using the mathematical model proposed by Zhao et al. (2014); the results are displayed in Fig. 3B. The estimated size of M. anatis core genome was approximately 649 genes. The analysis also revealed that M. anatis has an open pan-genome comprising more than 860 genes. In the Gram-negative mode bacteria Escherichia coli, the proportion of core genes in each genome is usually 42% (Zhao et al. 2014), which is much lower than that in M. anatis. This result also suggests that the gene pool of M. anatis is more conservative and the ability to obtain or lose genes is relatively weak.

Structure and function of the genic components in the M. anatis pan-genome. A The bar plot shows the number of gene clusters in the nth genome (n = 1, 2, … 16) of 16 M. anatis strains. B Estimation of pan- and core-genome sizes of M. anatis. The statistics for different genic groups in the nth genome were computed according to the datasets of 1,000 randomly selected strain combinations. C Functional profile of the M. anatis pan-genome. The color-coded bars denote core genes in magenta, dispensable genes in blue, and strain-specific genes in orange. The horizontal axis represents the COG functional class. The vertical axis represents the percentage of genes in each COG functional class. The details are summarized in Table S4. The proteins encoded by genes do not have homologs in the COG database and are not displayed. The asterisks denote significant enrichment of gene occurrence in the related COG category; the asterisks in black for the comparison between core and accessory genes; the asterisks in white for the comparison between core and strain-specific genes (FDR < 0.05; Fisher’s exact test) (Color figure online)
Next, we classified all the genes encoded by the pan-genome of M. anatis by COG functional classification. The results are summarized in Table S4 and are displayed in Fig. 3C. In some COG functional classes, the distribution of core genes is obviously different from that of dispensable genes and strain-specific genes. Eight COG functional classes were only found in core genes but deleted in dispensable genes and strain-specific genes; these included energy production and conversion, signal transduction mechanisms, intracellular trafficking and secretion, posttranslational modification, cell motility, nucleotide transport and metabolism, coenzyme transport and metabolism, and inorganic ion transport and metabolism. In addition, compared with dispensable genes and strain-specific genes, the protein products of core genes were significantly enriched in COG functional class Translation, ribosomal structure (FDR < 0.001; Fisher’s exact test). In contrast, compared to the set of dispensable genes, the two functional classes of defense mechanisms and mobilomes. Prophages and transposons had significantly fewer core genes (FDR < 0.05). The enrichment of translation-associated genes uniformly present in the genomes of all strains was consistent with their fundamental functions essential for bacterial growth and survival. In addition, nearly half (n = 375) of all pan-genome genes encoded the proteins assigned to the categories of “General function prediction only” and “Function unknown”, as well as the proteins without homologs in the COG collection. Due to mycoplasma is evolved from gram-positive bacteria through drastic genome reduction (Mushegian and Koonin 1996), the reduction of the genome means the loss of gene function and the loss of many biosynthetic capabilities, comparing with other bacteria. Through predictive COG analysis, it also confirmed the relatively simple structure and function, larger proportion of self-transport system and poor self-synthesis ability of mycoplasma from the genetic level. Compared with other avian mycoplasma, such as M.Columbinum, the distribution characteristics of metabolism-related core genes such as sugar、amino acid、nucleotide、coenzyme、lipid、inorganic ion transport and metabolism are basically similar, although there were large differences between their genome organizations. Interestingly, the core genes of sugar transport and related metabolic proteins of M. anatis is high, closed to genes related to DNA replication, recombination and repair. It implied that glycoproteins play an important role in the virulence and metabolism of M. anatis.
Distribution and structure of CRISPR-Cas
CRISPRs (clustered regularly interspersed palindromic repeats) are special arrays of repeat and spacer elements in the microbial genomes that often comprise DNA fragments derived from bacteriophages. CRISPRs play a vital role in antiviral defense systems of prokaryotes (Doron et al. 2018; Nethery and Barrangou 2019). Among our studied strains, CRISPRs were identified in the genomes of three strains (DP06, DP07, and DP12) but were absent in other M. anatis strains. As illustrated in Fig. 4, these CRISPRs were composed of 36-bp direct repeat (DR) elements individually separated by the spacers of 30 bp or occasionally 31 bp. Both CRISPRs of strains DP06 and DP07 had 23 copies of DRs, whereas four extra DRs were observed in DP12. Further BLASTn searching of the CRISPR arrays against CRISPRCasdb (Pourcel et al. 2020) revealed that some of the spacers in the M. anatis CRISPRs shared sequence similarities with the corresponding spacers from related bacteria (e.g., Salmonella enterica and Mahella australiensis) and archaea (e.g., Methanocaldococcus vulcanius and Sulfolobus islandicus). None of the spacer sequences from M. anatis CRISPRs were homologous to any phage or other known sequences in the NCBI database. Interestingly, the consensus DR sequence (GTCTATACTAACTCAACTTTAGCACAACCAAAAAAC) of M. anatis CRISPRs was unique compared with the known DR elements in CRISPRCasdb.

Comparison of the CRISPR-Cas systems in three M. anatis strains. The genetic organization of the CRISPR loci in green and their associated (Cas) genes in blue are shown in the upper panel. The other genes surrounding CRISPR loci are colored in grey. The CRISPR arrays of the repeat and spacer elements are illustrated in the bottom panel (Color figure online)
As is well known, a cluster of CRISPR-associated genes called cas usually surrounds CRISPR arrays, and together they constitute a CRISPR-Cas immune system to defend bacteria from the phage invasion (Jansen et al. 2002). Here, three CRISPR-associated genes cas1, cas2, and cas9 of M. anatis were identified in the vicinity of the CRISPRs detected above (Fig. 4). The cas genes together with the closely linked genes encoding hypothetical proteins were highly conserved in three strains (DP06, DP07, and DP12). Downstream of cas9, there was an operon containing cas1 and cas2 followed by a hypothetical protein transcribed in the opposite orientation. The CRISPR-associated endonucleases Cas9/2/1 of M. anatis shared 37%, 76%, and 71% amino acid identity, respectively, compared with the homologs of M. anserisalpingitis type strain ATCC BAA-2147 that is a goose pathogen (Grózner et al. 2019). It was worth noting that the genetic architecture of the CRISPR-Cas system of M. anatis was similar to those found in M. anserisalpingitis, M. hyosynoviae, and M. arthritidis but was different from the other Mycoplasma species (Grózner et al. 2019; Bumgardner et al. 2015; Bushnell B. 2014; Seemann T., 2014; Altschul et al. 1997; Pourcel et al. 2020). The role of the CRISPR-Cas system protecting the bacteria-degrading invaders has yet to be further verified for mycoplasmas.
Comparative analysis of virulence-associated genes
According to the analysis of virulence-related genes (VAGs) in the VF database, 29 genes encoding potential virulence factors were detected in the pan-genome of M. anatis. The genetic distribution and protein functions of these VAGs in 16 strains are shown in Fig. 5. Twenty-seven of the 29 VAGs were core genes, and they were highly conserved in all strains. According to the subcellular localization of proteins, these VAGs were divided into three categories, including 19 cytoplasmic membrane proteins, seven cytoplasmic proteins, and two proteins secreted into the extracellular space. In addition, there was a subcellular blurred protein that was classified as COG category "Cell wall/membrane/envelope biogenesis". Obviously, most of the identified M. anatis VAGs encoded membrane-associated proteins, and the functions of these proteins may be related to the bacterial pathogenicity, immunogenicity, and the interaction between pathogen and host.

Distribution of the genes with a potential role in virulence of M. anatis. The heatmap shows the presence and absence of the genes involved in biosynthesis of putative virulence factors across the strains of M. anatis. Protein subcellular localizations are color-coded for each gene, and the COG codes corresponding to individual functional category are recorded in Table S4
Bacterial ATP-binding cassette (ABC) transporters are often known to be important for cell viability and virulence (Davidson et al. 2008). Notably, more than half of the VAGs in the pan-genome of M. anatis coded for ATP-binding cassette (ABC) transporters, which are necessary for bacteria to absorb nutrients and secret specific toxins (Kaul and Pattan 2011). Some of these VAGs were found to be homologous to the ATPases of iron ABC uptake systems in other pathogenic bacteria, e.g., OG222/361/402/656 corresponding to the Haemophilus influenzae HitC, OG006/403 to the Neisseria meningitidis FbpC, and OG274 to the Escherichia coli FepC (Fig. 5). These iron transport proteins of M. anatis were predicted to be localized at the cytoplasmic membrane, where they may function in scavenging iron in host environments (Khun et al. 2000; Perkins-Balding et al. 2004). Additionally, six OGs (OG051/299/304/359/543/563) encoding ABC transporters were also determined to be cytoplasmic membrane proteins that were involved in mycoplasmal defense mechanisms (Fig. 5). These proteins share 49–53% sequence similarity with the lipid A flippase MsbA that contributes to lipid A export in H. influenzae. Interestingly, each of these defense-associated proteins possess two Pfam structure domains ABC_tran (PF00005) and ABC_membrane (PF00664), both of which are essential domains of a typical ABC transporter involved in the translocation of various substrates across the membrane, including sugars, peptides, and toxins (Razin 1998; Saurin et al. 1999).
Two VAGs (OG085/708) that were predicted to be secreted extracellular proteins shared 59% and 57% similarity, respectively, with the μ-toxin (hyaluronidase) NagH, a major component of Clostridium perfringens toxin complex (Vilhelmina and Gediminas Arvydas Biziulevi 2000). Notably, both proteins of M. anatis were also found to be carbohydrate-active toxins belonging to the glycoside hydrolase family 84 with three typical Pfam domains: NAGidase (PF07555), Glyco_hydro_20b (PF02838), and PF00754 (F5_F8_type_C) (Adams et al. 2008). Additionally, three genes (OG367/401/411/) coding for putative ATPase involved in the biogenesis of type III secretion system (T3SS) were identified in the pan-genome of M. anatis. T3SS has been characterized by its ability to deliver effectors into host cells during bacterial infections (Puhar and Sansonetti 2014).
Conclusions
This study, as the first report of the structure and function of the pan-genome of M. anatis, provides a valuable genetic basis for many aspects of future research on the pathogens of waterfowl. Our results are relevant to the molecular mechanisms of drug resistance, adaptive evolution of pathogens, population structure, and vaccine development. In addition, a complete virulence-related gene profile has been identified, and most of the genes are highly conserved in all strains; the data thus provide genetic markers for rapid diagnosis of clinical infection.
References
Adams JJ, Gregg K, Bayer EA, Boraston AB, Smith SP (2008) Structural basis of Clostridium perfringens toxin complex formation. Proc Natl Acad Sci USA 105(34):12194–12199
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al (1997a) Gapped BLAST and PSI BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477
Bencina D, Tadina T, Dorrer D (1988) Natural infection of ducks with Mycoplasma synoviae and Mycoplasma gallisepticum and Mycoplasma egg transmission. Avian Pathol 17(2):441–449
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC et al (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform 8(1):209
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinform 30(15):2114–2120
Bumgardner EA, Kittichotirat W, Bumgarner RE, Lawrence PK (2015) Comparative genomic analysis of seven Mycoplasma hyosynoviaestrains. Microbiol Open 4(2):343–359
Bushnell B (2014) BBMap: a fast, accurate, splice-aware aligner. Lawrence Berkeley National Lab (LBNL), Berkeley
Davidson AL, Dassa E, Orelle C, Chen J (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72(2):317–364
Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M et al (2018) Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359(6379):r4120
Grózner D, Forró B, Sulyok KM, Marton S, Kreizinger Z, Bányai K, Gyuranecz M (2018) Complete genome sequences of Mycoplasma anatis, M. anseris, and M. cloacale type strains. Microbiol Resour Announce 7(12):e00939-e1018
Grózner D, Forró B, Kovács RB, Marton S, Gyuranecz M (2019) Complete genome sequences of three Mycoplasma anserisalpingitis (Mycoplasma sp. 1220) Strains. Microbiol Resour Announce 8:e919–e985
Jansen R, van Embden JDA, Gaastra W, Schouls LM, Jansen R, van Embden J, Gaastra W, Schouls L (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575
Kaul G, Pattan G (2011) MsbA ATP-binding cassette (ABC) transporter of E. coli: Structure and possible flippase mechanism. Indian J Biochem Biophys 48(1):7–13
Khun HH, Deved V, Wong H, Lee BC (2000) fbpABC gene cluster in Neisseria meningitidis is transcribed as an operon. Infect Immun 68(12):7166–7171
Mushegian AR, Koonin EV (1996) A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA 93(19):10268–10273
Nethery MA, Barrangou R (2019) CRISPR Visualizer: rapid identification and visualization of CRISPR loci via an automated high-throughput processing pipeline. RNA Biol 16(4):577–584
Nguyen L, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(1):268–274
Page AJ, Cummins CA, Martin H, Wong VK, Sandra R, Holden MTG et al (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31(22):3691–3693
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) CheckM: assessing the quality ofmicrobial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055
Perkins-Balding D, Ratliff-Griffin M, Stojiljkovic I (2004) Iron transport systems in neisseria meningitidis. Microbiol Mol Biol Rev 68(1):154–171
Pourcel C, Touchon M, Villeriot N, Vernadet J-P, Couvin D, Toffano-Nioche C et al (2020a) CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and casgenes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res 48(D1):D535–D544
Puhar A, Sansonetti PJ (2014) Type III secretion system. Curr Biol. https://doi.org/10.1016/j.cub.2014.07.016
Razin S, Yogev D, Naot Y (1998) Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev 62(4):1094–1156
Richter M, Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci 106(45):19126–19131
Saurin W, Hofnung M, Dassa E (1999) Getting in or out: early segregation between importers and exporters in the evolution of ATP-binding cassette (ABC) transporters. J Mol Evol 48(1):22–41
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069
Stipkovits L, Kempf I (1997) Mycoplasmoses in poultry. Rev Sci Tech 15(4):1495–1525
Stipkovits L, Varga Z, Czifra G, Dobos-Kovács M (1986) Occurrence of mycoplasmas in geese affected with inflammation of the cloaca and phallus. Avian Pathol 15(2):289–299
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV et al (2003) The COG database: an updated version includes eukaryotes. BMC Bioinform 4:41
Vilei EM, Nicolet J, Frey J (1999) IS1634, a novel insertion element creating long, variable-length direct repeats which is specific for Mycoplasma mycoides subsp. mycoides small-colony type. J Bacteriol 181(4):1319–1323
Vilhelmina Z, Gediminas Arvydas Biziulevi C (2000) Physico-chemical and catalytic properties of Clostridium perfringens hyaluronidase: an update. Anaerobe 6(6):347–355
Waites KB, Xiao L, Liu Y, Balish MF, Atkinson TP (2017) Mycoplasma pneumoniae from the respiratory tract and beyond. Clin Microbiol Rev 30(3):747–809
Xie Z, Tang H (2017) ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics. 33(21):3340–7
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26(13):1608–1615
Zhao Y, Jia X, Yang J, Ling Y, Zhang Z, Yu J et al (2014) PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 30(9):1297–1299
Zhou Y, Yongjie L, Karlene HL, Jonathan JD, Wishart DS (2011) PHAST: a fast phage search tool. Nucl Acids Res 39(suppl):W347–W352
Funding
This work was partially supported by: Guangdong Basic and Applied Basic Research Fund (No. 2019B1515210008).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
13258_2021_1129_MOESM1_ESM.pdf
Figure S1. The heatmap of pairwise genome similarities color-coded by the ANI scores for the 16 M. anatis strains and the reference genomes of four Mycoplasma species as (PDF 70 kb)
13258_2021_1129_MOESM2_ESM.xlsx
Table S1. The quality statistics on genome completeness and contamination of 15 newly sequenced M. anatis strains (XLSX 10 kb)
13258_2021_1129_MOESM3_ESM.xlsx
Table S2. The genomic regions present in M. anatis strain LC01 but absent in strain NCTC 10156. The sequence fragments greater than 300 bp are shown (XLSX 10 kb)
13258_2021_1129_MOESM5_ESM.xlsx
Table S4. Summary data of gene clustering for all protein-coding sequences encoded in 16 M. anatis genomes (XLSX 147 kb)
13258_2021_1129_MOESM6_ESM.xlsx
Table S5. Summary of COG functional classification for the core, dispensable, and strain-specific genes in the pangenome of M. anatis (XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Zhou, Q., Mai, K., Yang, D. et al. Comparative genomic analysis of Mycoplasma anatis strains. Genes Genom 43, 1327–1337 (2021). https://doi.org/10.1007/s13258-021-01129-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-021-01129-5