
Abstract
Although surface-enhanced Raman spectroscopy (SERS) is a powerful analytical technique with unbeaten sensitivity, the capabilities of SERS have been not fully utilized in screening applications because throughput of spectrum detection by conventional Raman instruments has been restricted due to their single-point measurement manners. Hence, this paper presents a development of a high throughput Raman screening system that employs a fiber-optic switch and a Raman probe array. In the system, a 785 nm excitation light is directed into the 1 × 8 broadband optical switching device and selectively switched to one of 8 output ports connected to the corresponding Raman probe array to deliver the light to samples under each probe. This optical switching driven probing in sequence allows us to rapidly detect Raman scattering of the multiple (n = 8) samples in array within a short time (~ 28 s) with decent sensitivity (10–7 M). The Raman spectroscopy of the system is validated by comparing the features of Raman spectra obtained from vitamin C tablets with those from a commercial Raman microscope and the detection sensitivity is measured with SERS substrates with different concentrations. Then, feasibility of high throughput screening is tested with a SERS chip array.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Raman spectroscopy is a light scattering based chemical analysis technique. It interrogates an inelastic light scattering, known as Raman scattering in which photons incident on a sample lose or gain energy via interaction with vibrating molecules in the illuminated sample [1]. This energy shift is spectrally encoded in the detected Raman scattered photons of which spectra reflect information regarding molecular compositions of the sample. Thus, Raman spectroscopy can be applied to detect characteristic chemical fingerprints of versatile substances in any phase (gas, solid, and liquid) without sample labeling, allowing a wide range of applications for quantitative chemical analysis, including blood and drug analysis [2, 3]. Despite of the advantages, wide-spread application of Raman spectroscopy in real-life detection has been limited because the normal Raman scattering is intrinsically faint and occurs for one in every 108 scattered photons [4].
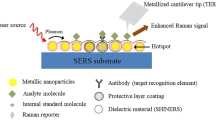
Surface-enhanced Raman spectroscopy (SERS) is an alternative to overcome the limitation of Raman spectroscopy [5]. SERS detects surface enhanced Raman scattering, a phenomenon in which inelastic light scattering by molecules is greatly enhanced (by factors up to 108 or even larger) when the molecules are absorbed onto corrugated metallic nanostructures [6]. The enormous enhancement in Raman intensity is mainly attributed to the excitation of the localized surface plasmons and the chemical interactions [7, 8]. More specifically, as the metal nanostructures interact with an electromagnetic wave (incident light), the collective oscillations of surface electrons (well known as surface plasmon) are induced, generating very large electric fields occurred at the sharp apexes and edges in the illuminated nanostructures [9]. Chemical enhancement arises from charge transfer between the plasmonic nanostructure and the analyte molecules in the vicinity of the nanostructures, resulting in the enhancement of Raman scattering [10]. Owing to its ultra-high sensitivity, SERS allows for the structural fingerprinting of low concentration analytes. The recent technical advances in SERS have reported the achievable enhancement factor as large as 1014 [11], suggesting the way to detection on the single-molecule level while preserving the main advantages of parent Raman spectroscopy. Therefore, SERS is considered a very promising option for routine analytical techniques in vast array fields including biochemical, biomedical, environmental, and food analyses [12,13,14,15,16,17].
In screening applications, to fully leverage the powerful capabilities of SERS, it is essential to improve the throughput in spectrum detection of the Raman systems. However, the throughput of the conventional Raman microscopes has been highly restricted due to their single point measurement manner, and thus it requires intensive human labor for testing of multiple samples, resulting in long screening times of more than tens of minutes to hours for tens of samples [18, 19]. However, high throughput screening (HTS) has been used as a key technique to analyze the number of chemical compounds in the recent pharmaceutical industry and biomedical researches [20, 21]. In HTS, a combination of microplates containing 96- or 384-wells and plate readers allows for rapid testing of tens to hundreds of chemical, genetic, or biological libraries [22]. Hence, the combination of a high throughput screening with Raman spectroscopy can be effective for the rapid SERS detection of massive samples. While the HTS-Raman systems have been developed and commercially available [23, 24], they still adhere the single-point, one-by-one detection manners using a slow mechanical x–y translation stage although it runs automatically, which may restrict the high throughput SERS detection in practice. Recently, Kawagoe et al. have successfully demonstrated parallel Raman detection of hundreds of samples in the multi-well plate [25]. Despite of significant improvement in throughput of Raman measurement, however, it needs the hundreds of customized Raman probes facing each well for simultaneous detection and high priced two-dimensional (2D) spectrometer, making it complex and costly.
In this paper, we develop an optical switch based Raman spectroscopy (OSRS) system for high throughput screening that combines a fiber-based optical switching device and a Raman probe array in a confocal Raman microscope. The optical switch has been employed as a useful multi-port optical networking connector in the telecommunication industry owing to its high speed and low-loss optical power delivering capability on a broad bandwidth [26]. In the proposed system, the fiber based optical switch is exploited to selectively switch the Raman probes. In the device, the input port is mechanically linked to one of the output ports connected to the corresponding probes, enabling sequential light transport to each probe in few milliseconds. Thus, the high-speed optical switching manner allows us to rapidly detect Raman spectra of the multi-samples in array, useful for massive workload assay. The system performance as Raman spectroscopy is validated by measuring Raman spectra of vitamin C tablets and different SERS substrates. Then, the system feasibility for high throughput screening is demonstrated with a SERS chip array.
2 Materials and Methods
2.1 Optical-Switch Raman Spectroscopy
Figure 1a shows a schematic of the implemented OSRS setup. The system consists of three main components: a light source, an array probe with an optical switch, and a spectrometer. A 785-nm laser diode (I0785SL0100SA, IPS Inc.) with a spectral linewidth of 0.2 nm was used as an excitation light source, and the output beam from the laser was collimated by passing it through a collimator (TC25FC-780, Thorlabs Inc.). The power of the collimated beam was adjusted using a circular variable neutral-density (ND) filter (NDC-100C-2-B, Thorlabs Inc.). The beam was deflected 90° by a dichroic mirror (LPD02-785RU-25, transmission at > 785 nm, Semrock Inc.) and then directed to an input fiber of a broad wavelength mini optical switch (FFMU-088S1H229, Agiltron Inc.) via another collimator (TC25FC-780, Thorlabs Inc.). The optical switch used has 1 × 8 optical channels connected to 0.5-m-long optical fibers with a core diameter of 400 μm, among which one was used as the input optical channel and the remaining fibers were used as the output optical channels. The input optical channel was connected to each output optical channel by directly coupling a pair of fibers. This was achieved by moving the input optical fiber using a precision mini-motor in the switch so that the beam directed to the input channel could be transferred to the coupled output fiber. The location of the input fiber was adjusted by the driving circuit, which interfaced with a PC through a USB connection. Additionally, the eight output fibers were connected to eight identical small-lens optics (numerical aperture: 0.64, focal length: 4.03 mm) assembled onto an aluminum plate as a multi-array probe. The beam from the probe was tightly focused on the sample, which was placed on a translation stage, and the scattered light (Raman + Rayleigh scattering) from the sample was collected from the output fiber and redirected to the input fiber. The light signals with wavelengths exceeding 785 nm were passed through the dichroic filter. The transmitted light signal (Raman signal) was spectrally filtered by two long-pass filters (LP02-785RE-25, Semrock Inc.) to ensure the suppression of the residual Rayleigh scattering light. Finally, the filtered Raman signal was detected by a highly sensitive portable spectrometer (MNL-1009 QE Pro, Ocean Insight Inc.) to display the Raman spectrum. The system operation (optical switching, signal detection, display, and storage) was fully automated by a customized operating program written in LabVIEW. Figure 1b shows a photograph of the OSRS system implemented in the laboratory.

a Schematic of the confocal Raman spectroscopy equipped with a fiber-based optical switch and b photograph of the implemented system
2.2 Raman Data Acquisition
The overall procedure for Raman data acquisition using the optical switching is illustrated as a flow chart in Fig. 2a. In briefly, the spectrometer and optical switch are activated and initiated, during which the input optical fiber in the optical switch is physically situated to the first (i = 1) output optical The overall procedure for Raman data acquisition using optical switching is illustrated in Fig. 2a. Briefly, the spectrometer and optical switch are activated and initiated, during which the input optical fiber in the optical switch is physically placed at the first (i = 1) output optical channel (“home” channel, corresponding to ready mode). Next, for optical switching, a command to connect the input optical fiber to the next (i = 2) output optical channel is sent to the driving circuit of the optical switch, where the input fiber is moved and coupled to the adjacent output fiber by the mini-motor in 500 ms. The completion of the channel switching is confirmed by a Boolean indicator in the system operating program (lighting of a green lamp in Fig. 2b). Immediately after the green lamp turns on, the Raman signal from the probe connected to the i-th output optical fiber is detected by the spectrometer and displayed (Fig. 2b). If the current i is not equal to 8 (total number of output channels), i is increased to i + 1 (i = 3), and the channel conversion of the neighboring output fiber, followed by the Raman spectra detection and display, is repeated until i is 8. Thus, Raman data from the eight samples in the array are sequentially acquired and displayed within a few seconds (total switching time is 4 s + integration time depending on the samples) (Fig. 2c). The acquired Raman dataset is saved in a CSV format on the PC for analysis. These procedures (channel switching, data acquisition, and display) are shown in Video S1.

a Flowchart of how Raman data are acquired using the OSRS system. b Sequence for channel switching and data acquisition, corresponding to the blue dotted box in (a). c Front panel of the LabVIEW operating program (top: before measurement, bottom: after measurement), with which eight monitors sequentially display the Raman spectrum for each channel (channels 1–8)
2.3 Raman Data Calibration
The Raman data measured by our system can be affected by two strong background signals. The first is a fiber background (FB) signal due to Raman scattering coming from the fiber core material (silica) as the laser beam propagates along the fiber channels in the optical switch [27]. The other is a fluorescence signal emitted from the sample excited by the laser beam. These signals are comparable to or even stronger than the sample Raman signal and can cause a misleading readout of the actual sample’s Raman data. Thus, to remove the background signals, the acquired Raman data are corrected via two simple numerical steps, as explained in the following paragraph.
The first step is to utilize the sample-free FB signal to remove the FB signal from the sample data. The sample-free FB signal is the fiber’s Raman signal acquired with no sample, and its spectrum is static. We found that the peaks of this Raman signal are dominant even with the sample. Considering that the Raman signal intensity is positively correlated with the laser power and integration time [28], the FB signal from the sample data is removed by rescaling the intensities of the Raman peaks of the sample-free FB signal to those of the sample data and then subtracting them. For example, Fig. 3a shows the sample-free FB signal (red) and the sample Raman data (black). From the FB signal, we selected the main peak at 487 cm−1 as a reference peak for rescaling, which is distinct and far from the sample spectra (600–1800 cm−1). Then, the FB signal was numerically rescaled with a scale factor (intensity ratio of the peaks at 487 cm−1) as follows:
where IRescaled FB represents the rescaled FB signal (red in Fig. 3b) and IFB represents the original FB signal before rescaling (black in Fig. 3b). As shown in Fig. 3b, the rescaled FB signal is well-matched with the FB signal in the sample Raman data. Thus, after the subtraction of the rescaled FB signal, the sample’s Raman spectrum remains on the broad and smooth emission spectrum resulting from the fluorescence of the sample, as shown in Fig. 3c. The emission spectrum is then easily removed via polynomial fitting as a second step (Fig. 3d), yielding the Raman spectrum of only the sample (Fig. 3e).

Numerical calibration of the sample Raman data to remove Raman signal of fiber and emission spectrum due to sample fluorescence
3 Results and Discussion
3.1 Raman Signal Measurement
The performance of the proposed system for Raman spectroscopy was tested with chemical compounds and SERS chips. We selected a vitamin C (ascorbic acid) tablet as a testing sample because it is well-standardized in size and chemicals, easy to align and probe. Moreover, Raman spectrum of the vitamin C has been commonly reported in many previous literatures [29, 30], which can be used as a reference for Raman spectra obtained by the proposed system. The 785-nm laser beam was focused on the tablet with an optical power of 8.2 mW for 3 s. Figure 4a shows the Raman spectrum of vitamin C measured by one of the eight probes in the OSRS system, which was spectrally analogous to the Raman signal of the same sample measured by a commercial confocal Raman microscope (HEDA, Weve Inc.) with the similar experimental condition (laser wavelength: 785 nm, laser power: 8 mW, lens NA: 0.64, exposure time: 3 s) The spectral shape in Fig. 4a was consistent with those reported in the previous Raman spectroscopy papers [29, 30], where the peaks at 623 cm−1, 1122 cm−1, and 1656 cm−1 are assigned to the C–C, C–O–C, and C = C stretching vibrations, respectively. Moreover, the peaks at 1257 cm−1 and 1315 cm−1 are attributed to the C–O–H and CH bending bonds, respectively. These results indicate that the Raman signal was detected by the OSRS system.

Raman spectroscopy demonstrating system performance. a Raman spectra of a vitamin C tablet measured by the proposed OSRS system and a commercial Raman microscope system under the same conditions (beam power: 8.2 mW, integration time: 3 s). b Three-dimensional plot of the Raman spectra for vitamin C tablets detected by all (n = 8) optical switching probes of the OSRS system. c Intensity profiles obtained from the main spectral peaks (at 623 cm−1, 1121 cm−1, 1256 cm−1, 1315 cm−1, and 1657 cm−1) in (b)
Serial measurements were conducted for all (n = 8) probes using the optical switch. The data acquisition took ~ 28 s, including the switching time (0.5 s) and integration time (3 s) for each channel. Figure 4b presents the Raman spectra of the vitamin C tablets placed under each probe, showing similarities in terms of the spectrum and magnitude despite minor differences in the intensities of the main peaks at 623 cm−1, 1121 cm−1, 1256 cm−1, 1315 cm−1, and 1657 cm−1 (Fig. 4c). This small ripple in the intensities may be due to sample misalignment or the non-flat sample stage inducing different focal beam powers onto the samples. However, the results indicate uniform performance in Raman detection for all channels.
3.2 System Sensitivity Measurement
To evaluate the SERS signal sensitivity of our system, we used four well-estimated SERS chips based on an Au-nanoparticle-internalized nanodimple substrate [31], which was freely donated. To generate the SERS output of the substrate, malachite green isothiocyanate (MGITC), which is commonly used as a surface-bound Raman reporter, was topically applied to the SERS chips. In our experiment, four SERS chips with different MGITC concentrations (ranged from 10–7 M to 10–4 M) were fabricated, and their SERS signals were measured using the OSRS system. The SERS spectra for the MGITC solutions of different concentrations are shown in Fig. 5a. As the MGITC concentration increased, the intensity of the SERS signal increased. The main Raman peaks of the SERS spectra at 915 cm−1, 1171 cm−1, 1293 cm−1, 1363 cm−1, 1391 cm−1, and 1582 cm−1 are attributed to the ring-breathing, in-plane benzene v9, phenyl-N + C–C stretching, in-plane C–C and C-H stretching, and in-plane phenyl ring stretching and bending vibrations, respectively [32, 33]. Most of the six peaks were observable for low concentrations of 10–7 M. The SERS intensities at 915 cm−1, 1171 cm−1, 1391 cm−1, and 1582 cm−1 were plotted with respect to the MGITC concentration (Fig. 5b). High linearity between the SERS intensity and MGITC concentration was observed above 10–6 M for all the peaks (R2 = 0.9726 at 915 cm−1, 0.9913 at 1171 cm−1, 0.9899 at 1391 cm−1, and 0.97 at 1582 cm−1). The result shows that the system can provide the SERS measurements in analyte concentrations as low as 10–7 M, representing the detection sensitivity.

a SERS signals of MGITC with different concentrations in the range of 10–7–10–4 M measured by the OSRS system. b SERS intensity plots for 915 cm−1, 1171 cm−1, 1391 cm−1, and 1582 cm−1. The error bars indicate the standard deviation of 10 measurements of the SERS active sites on the chips
3.3 SERS Measurement
To perform the SERS measurements of various analytes, five chemical reagents were used: Rhodamine B (RhB, Junsei Chemical), Rhodamine 6G (R6G, Sigma–Aldrich), malachite green chloride (MG, Supelco Inc.), 1,2-di(4-pyridyl) ethylene (BPE, Sigma–Aldrich), and 3,3′-diethylthiadicarbocyanine iodide (DTDCI, Sigma–Aldrich). The RhB (479 μg/mL), R6G (479 μg/mL), and DTDCI (5.18 μg/mL) powders were diluted in deionized water, and the MG (364.9 μg/mL) and BPE (182 μg/mL, 5% methanol in water as solvent) powders were synthesized in methanol at a concentration of 1 mM. Next, the compounds were dropped (10 μL of solution) onto SERS chips with an Au-coated zinc oxide (ZnO) vertical nanorod substrate [34] and evaporated via the drop-casting method prior to the SERS measurements using the OSRS system. The SERS signals were obtained with a 10-mW laser power for 3 s. The unique SERS patterns of the respective molecules can be clearly distinguished by their narrow bands, as shown in Fig. 6a, and are similar to those previously reported [35,36,37,38,39]. In the RhB spectrum, the peaks at 1280 cm−1, 1356 cm−1, and 1507 cm−1 are attributed to the C–C bridge stretching and aromatic stretching [35]. The bands at 1359 cm−1 and 1507 cm−1 are from aromatic C–C stretching in R6G [36]. The peaks at 1174 cm−1, 1397 cm−1, and 1615 cm−1 in the MG spectrum are assigned to the in-plane vibrations of the C–H ring, N-phenyl stretching, and C–C ring stretching, respectively [37]. The peaks at 1199 cm−1, 1606 cm−1, and 1638 cm−1 in the BPE spectrum correspond to the in-plane vibrations of the C-H ring, pyridyl ring C–C stretching, and vinyl C=C stretching, respectively [38]. To visually identify the spectral characteristics of the sample, the spectrum is presented as a pseudo-colormap in Fig. 6b. The colors indicate the normalized magnitudes of the spectral intensities; the hotter color regions correspond to the main Raman peaks (red-shaded bars) of the samples in Fig. 6a. The results indicate that the proposed system can detect and identify the SERS signals of the different compounds.

a SERS signals of five types of Raman labeling compounds (RhB, R6G, MG, BPE, and DTDCI) that were deposited on the SERS chips. b Pseudo-colormaps of the Raman spectral intensities in (a), where the hot narrow bands in the map correspond to the main peaks of the Raman spectrum
3.4 SERS Array Measurement
We evaluated the massive testing feasibility of the system by fabricating an SERS array sensor (152 mm × 52 mm) comprising 24 SERS chips uniformly arranged in a 3 × 8 (rows × columns) two-dimensional (2D) array on silica glass. One of the five reagents (DTDCI, BPE, MG, R6G, and RhB) in Fig. 6 was randomly selected and deposited on the individual SERS chips. Then, the fabricated 24 SERS chips were arranged with 8 chips per row on a three-axis translation stage under the Raman probes of the OSRS system, as shown in Fig. 7a. The distance between adjacent SERS chips was equal to the pitch of the Raman probe array (17 mm), allowing the laser beam to be focused on the center of each chip’s substrate. The SERS measurement for the 1st line was conducted by sweeping the laser beam along the Raman probes, after which the sensor was forward-shifted for the next line measurement by using the translation stage. The total measurement time for each line was 28 s. Figure 7b–d present the colormaps of the Raman spectra measured from the SERS chip array in the 1st, 2nd, and 3rd lines, respectively. The colormaps exhibit the characteristic sample patterns of the Raman bands, distinguishing the reagents on the SERS chips. This result indicates that the proposed Raman system can rapidly detect and identify multi-variable single target molecules on metallic nanostructures.

a Photograph of the 3 × 8 SERS chip array sensor on the sample stage of the OSRS system. b–d Colormaps showing the Raman spectra of the SERS signals from eight SERS chips for the 1st, 2nd, and 3rd lines of the sensor, respectively
In discussion, the measurement time of the proposed system was ~ 28 s including the data acquisition and display for the multiple samples (n = 8) in array, corresponding to the screening time of ~ 3.5 s (0.5 s switching time + 3 s integration time) per sample. Since the screening is mainly limited to the integration time, the measurement time can be significantly shortened to 12 s for 8 samples by decreasing the exposure time to 1 s at the expense of the intensity levels. Furthermore, the switching time can be also decreased by using digital MEMS-driven fiber optic switches, in which the on/off switching between output ports is achieved by thermally activated digital micro-mirror movements within 10 ms [40]. For a standard 96-well plate, therefore, the system is expected to screen all wells (n = 96) within a few minutes considering the mechanical translation of the well plate, which is much faster compared to the conventional Raman microscopes (> several tens of minutes for tens of samples) [25].
In this work, the detection sensitivity of proposed system was estimated to be 10–7 M, the lowest concentration in the MGITC SERS substrates provided, which is comparable to the previous results obtained using the commercial Raman systems [41]. Improvement in the sensitivity (less than 10–7 M) would be possible with high quality SERS substrates with a very low detection limit (LOD) for specific molecules [42, 43]. Furthermore, the use of lens with the higher NA more than 0.6 (the corresponding magnification is typically 40 ~ 100) may increase the Raman signal levels for the sample optical power, compromising of the shorter working distance [44].
We have observed the strong Raman spectral background generated in the silica cores of the excitation and collection fibers in the fiber-optic switch and probe, which could be effectively removed from the measured spectra using the simple numerical method (Fig. 3). There have been the technical approaches to avoid the high silica background. Instead of the lone fiber used for both excitation laser delivery and Raman signal collection, multiple, bundled silica fibers have been used in the fiber-optic probe to separate the excitation and collection paths [45, 46]. Konorov et al. have proposed a multi-fiber probe based on a hollow-core photonic-crystal fiber (PCF) surrounded by three silica-core collection fibers [47]. Because the excitation beam is transmitted through air hole (10–20 μm in diameter) inside the hollow-core of PCF, the silica Raman scattering can be much reduced. However, the multi-fiber configuration may lead to the increased size and complexity of the system. Raml et al. have demonstrated Raman spectroscopy using a single crystal sapphire fiber [48]. Because of its narrow Raman bandwidth close to the excitation laser wavelength, the background Raman spectrum remains localized, and the sample Raman spectra can be easily identified.
The proposed system may be also utilized for high throughput fluorescence based immunoassay [49]. To achieve both SERS and immunoassay on the single platform, the modification in the current system is considered. For example, two single wavelength lasers (785 nm laser for SERS, visible laser for immunoassay) would be used as excitation light sources for Raman scattering and fluorescence emission [50], respectively. Moreover, a wavelength division multiplexer (WDM) combiner would be needed to deliver the two laser beams into the optical switch for sample illumination, and an additional optical component such as a broadband beam splitter would be placed between the WDM and the Raman dichroic mirror to deflect the fluorescence emission signals. In the detection part, a photomultiplier tube (PMT) would be added for the fluorescence signal detection.
4 Conclusion
We have developed a fiber-optic switch based Raman spectroscopy to tackle the limitation in throughput of conventional Raman instruments. Our experiments demonstrated its feasibility for high throughput screening of multi-variable chemical molecules. The developed Raman system can be further improved in terms of the form factor, analytical capability, and full automation for its application to clinical and point-of-care settings, being currently underway as a near future work. With the SERS sensors, therefore, the proposed approach would be an alternative tool for high throughput Raman screening of SERS-active samples, including DNA, microRNA, proteins, blood, and bacteria.
Data availability
Not applicable.
References
Shipp, D., Sinjab, F., Notingher, I.: Raman spectroscopy: techniques and applications in the life sciences. Adv. Opt. Photonics 9, 315–428 (2017). https://doi.org/10.1364/AOP.9.000315
Atkins, C.G., Buckley, K., Blades, M.W., Turner, R.F.B.: Raman spectroscopy of blood and blood components. Appl. Spectrosc. 71, 767–793 (2017). https://doi.org/10.1177/0003702816686593
West, M.J., Went, M.J.: Detection of drugs of abuse by Raman spectroscopy. Drug Test Anal. 3, 532–538 (2010). https://doi.org/10.1002/dta.217
Jones, R.R., Hooper, D.C., Zhang, L., Wolverson, D., Valev, V.K.: Raman techniques: fundamentals and frontiers. Nanoscale Res. Lett. 14, 231 (2019). https://doi.org/10.1186/s11671-019-3039-2
Ding, S.-Y., Yi, J., Li, J.-F., Ren, B., Wu, D.-Y., Panneerselvam, R., Tian, Z.-Q.: Nanostructure-based plasmon-enhanced Raman spectroscopy for surface analysis of materials. Nat. Rev. Mater. 1, 16021 (2016). https://doi.org/10.1038/natrevmats.2016.21
Langer, J., et al.: Present and future of surface-enhanced raman scattering. ACS Nano 514, 28–117 (2020). https://doi.org/10.1021/acsnano.9b04224
Lombardi, J.R., Birke, R.L.: A unified view of surface-enhanced Raman scattering. Acc. Chem. Res. 42, 734–742 (2009). https://doi.org/10.1021/ar800249y
Moskovits, M.: Surface roughness and the enhanced intensity of Raman scattering by molecules absorbed on metals. J. Chem. Phys. 69, 4159–4161 (1978). https://doi.org/10.1063/1.437095
Willets, K.A., Van Duyne, R.P.: Localized surface plasmon resonance spectroscopy and sensing. Annu. Rev. Phys. Chem. 58, 267–297 (2007). https://doi.org/10.1146/annurev.physchem.58.032806.104607
Valley, N., Greeneltch, N., Van Duyne, R.P., Schatz, G.C.: A look at the origin and magnitude of the chemical contribution to the enhancement mechanism of surface-enhanced Raman spectroscopy (SERS): theory and experiment. J. Phys. Chem. Lett. 4, 2599–2604 (2013). https://doi.org/10.1021/jz4012383
Le Ru, E.C., Blackie, E., Meyer, M., Etchegoin, P.G.: Surface enhanced raman scattering enhancement factors: a comprehensive study. J. Phys. Chem. C 111, 13794–13803 (2007). https://doi.org/10.1021/jp0687908
Pyrak, E., Krajczewski, J., Kowalik, A., Kudelski, A., Jaworska, A.: Surface enhanced Raman spectroscopy for DNA biosensors-how far are we? Molecules 24, 4423 (2019). https://doi.org/10.3390/molecules24244423
Lee, W.-J., Kim, K.-J., Hossain, M.K., Cho, H.-Y., Choi, J.-W.: DNA-gold nanoparticle conjugates for intracellular miRNA detection using surface-enhanced Raman spectroscopy. BioChip J. 16, 33–40 (2022). https://doi.org/10.1007/s13206-021-00042-z
Lee, S., et al.: Nano-biomarker-based surface-enhanced Raman spectroscopy for selective diagnosis of gallbladder and liver injury. BioChip J. 16, 49–57 (2022). https://doi.org/10.1007/s13206-022-00045-4
Choi, N., Dang, H., Das, A., Sim, M.S., Chung, I.Y., Choo, J.: SERS biosensors for ultrasensitive detection of multiple biomarkers expressed in cancer cells. Biosens. Bioelectron. 164, 112326 (2020). https://doi.org/10.1016/j.bios.2020.112326
Ong, T.T.X., Blanch, E.W., Jones, O.A.H.: Surface enhanced Raman spectroscopy in environmental analysis, monitoring and assessment. Sci. Total Environ. 720, 137601 (2020). https://doi.org/10.1016/j.scitotenv.2020.137601
Jiang, L., Hassan, M.M., Ali, S., Li, H., Sheng, R.: Evolving trends in SERS-based technique for food quality and safety: a review. Trends Food Sci. Technol. 112, 225–240 (2021). https://doi.org/10.1016/j.tifs.2021.04.006
Kojima, T., Tsutsumi, S., Yamamoto, K., Ikeda, Y., Moriwaki, T.: High-throughput cocrystal slurry screening by use of in situ Raman microscopy and multi-well plate. Int. J. Pharm. 399, 52–59 (2010). https://doi.org/10.1016/j.ijpharm.2010.07.055
Jones, L.E., Stewart, A., Peter, K.L., McNaul, M., Speers, S.J., Fletcher, N.C., Bell, S.E.J.: Infrared and Raman screening of seized novel psychoactive substances: a large scale study of >200 samples. Analyst 141, 902–909 (2016). https://doi.org/10.1039/C5AN02326B
Macarron, R., et al.: Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 10, 188–195 (2011). https://doi.org/10.1038/nrd3368
Mishra, K.P., Ganju, L., Sairam, M., Banerjee, P.K., Sawhney, R.C.: A review of high throughput technology for the screening of natural products. Biomed. Pharmacother. 62, 94–98 (2008). https://doi.org/10.1016/j.biopha.2007.06.012
Mayer, L.M., Bojanic, D.: Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 9, 580–588 (2009). https://doi.org/10.1016/j.coph.2009.08.004
Medipally, D.K.R., Maguire, A., Bryant, J., Armstrong, J., Dunne, M., Finn, M., Lyng, F.M., Meade, A.D.: Development of a high throughput (HT) Raman spectroscopy method for rapid screening of liquid blood plasma from prostate cancer patients. Analyst 142, 1216–1226 (2017). https://doi.org/10.1039/C6AN02100J
https://optosky.com/all-auto-high-throughput-portable-raman-analyzer.html.
Kawagoe, H., Ando, J., Asanuma, M., Dodo, K., Miyano, T., Ueda, H., Sodeoka, M., Fujita, K.: Multiwell Raman plate reader for high-throughput biochemical screening. Sci. Rep. 11, 15742 (2021). https://doi.org/10.1038/s41598-021-95139-8
Marom, D.M., Miyamoto, Y., Neilson, D.T., Tomkos, I.: Optical switching in future fiber-optic networks utilizing spectral and spatial degrees of freedom. Proc. IEEE 110, 1835–1852 (2022). https://doi.org/10.1109/JPROC.2022.3207576
Hollenbeck, D., Cantrell, C.D.: Multiple-vibrational-mode model for fiber-optic Raman gain spectrum and response function. J. Opt. Soc. Am. B 19, 2886–2892 (2002). https://doi.org/10.1364/JOSAB.19.002886
McCreery, R.L.: Raman spectroscopy for chemical analysis. John Wiley, Sons, Inc., Hoboken (2000)
Yang, H., Irudayaraj, J.: Rapid determination of vitamin C by NIR, MIR, and FT-Raman techniques. J. Pharm. Pharmacol. 54, 1247–1255 (2002). https://doi.org/10.1211/002235702320402099
Panicker, C.Y., Varghese, H.T., Philip, D.: FT-IR, FT-Raman and SERS spectra of Vitamin C. Spectrochim. Acta A Mol. Biomol. Spectrosc. 65, 802–804 (2006). https://doi.org/10.1016/j.saa.2005.12.044
Dang, H., Park, S.-G., Wu, Y., Choi, N., Yang, J.-Y., Lee, S., Joo, S.-W., Chen, L., Choo, J.: Reproducible and sensitive plasmonic sensing platforms based on Au-nanoparticle-internalized nanodimpled substrates. Adv. Funct. Mater. (2021). https://doi.org/10.1002/adfm.202105703
Huang, C.-Y., Chien, C.-H.: Facile fabrication of micro/nano hierarchical SERS sensor via anisotropic etching and electrochemical treatment for malachite green detection. Appl. Sci. 9, 5237 (2019). https://doi.org/10.3390/app9235237
Leng, W., Vikesland, P.J.: MGITC facilitated formation of AuNP multimers. Langmuir 30, 8342–8349 (2014). https://doi.org/10.1021/la501807n
Jue, M., et al.: Optimization of ZnO nanorod-based surface enhanced raman scattering substrates for bio-applications. Nanomaterials 9, 447 (2019). https://doi.org/10.3390/nano9030447
Zhang, L., Li, P., Luo, L., Bu, X., Wang, X., Zhao, B., Tian, Y.: Sensitive detection of rhodamine b in condiments using surface-enhanced resonance Raman scattering (SERRS) silver nanowires as substrate. Appl. Spectrosc. 71, 2395–2403 (2017). https://doi.org/10.1177/0003702817711700
He, X.N., Gao, Y., Mahjouri-Samani, M., Black, P.N., Allen, J., Mitchell, M., Xiong, W., Zhou, Y.S., Jiang, L., Lu, Y.F.: Surface-enhanced Raman spectroscopy using gold-coated horizontally aligned carbon nanotubes. Nanotechnology 23, 205702 (2012). https://doi.org/10.1088/0957-4484/23/20/205702
Jin, Y., Ma, P., Liang, F., Gao, D., Wang, X.: Determination of malachite green in environmental water using cloud point extraction coupled with surface-enhanced Raman scattering. Anal. Methods 5, 5609–5614 (2013). https://doi.org/10.1039/C3AY41128A
Zhuang, Z., Shi, X., Chen, Y., Zuo, M.: Surface-enhanced Raman scattering of trans-1,2-Bis (4-Pyridyl)-ethylene on silver by theory calculations. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 79, 1593–1599 (2011). https://doi.org/10.1016/j.saa.2011.05.017
Huang, H., et al.: Competitive reaction pathway for site-selective conjugation of Raman dyes to hotspots on gold nanorods for greatly enhanced SERS performance. Small 10, 4012–4019 (2014). https://doi.org/10.1002/smll.201400860
https://agiltron.com/product/mems-1x8-latching-type-optical-switch/.
Kim, K., Choi, N., Jeon, J.H., Rhie, G.-E., Choo, J.: SERS-based immuoassays for the detection of botulinum toxins A and B using magnetic beads. Sensors 19, 4081 (2019). https://doi.org/10.3390/s19194081
Tzeng, Y., Lin, B.-Y.: Silver SERS adenine sensors with a very low detection limit. Biosensors 10, 53 (2020). https://doi.org/10.3390/bios10050053
Shahine, I., Mevellec, J.-Y., Richard-Plouet, M., Humbert, B., Tessier, P.-Y.: Nanoporous gold stacked layers as substrates for SERS detection in liquids or gases with ultralow detection limits and long-term stability. J. Phys. Chem. C 126, 17223–17233 (2022). https://doi.org/10.1021/acs.jpcc.2c05903
Tuschel, D.: The effect of microscope objectives on the Raman spectra of crystals. Spectroscopy 32, 14–23 (2017)
Motz, J.T., Hunter, M., Galindo, L.H., Gardecki, J.A., Kramer, J.R., Dasari, R.R., Feld, M.S.: Optical fiber probe for biomedical Raman spectroscopy. Appl. Opt. 43, 542–554 (2004). https://doi.org/10.1364/AO.43.000542
Petterson, I.E.I., Day, J.C.C., Fullwood, L.M., Gardner, B., Stone, N.: Characterisation of a fibre optic Raman probe within a hypodermic needle. Anal. Bioanal. Chem. 407, 8311–8320 (2015). https://doi.org/10.1007/s00216-015-9021-7
Konorov, S.O., Addison, C.J., Schulze, G., Turner, R.F.B., Blades, M.W.: Hollow-core photonic crystal fiber-optic probes for Raman spectroscopy. Opt. Lett. 31, 1911–1913 (2006). https://doi.org/10.1364/OL.31.001911
Raml, C., He, X., Han, M., Alexander, D.R., Lu, Y.: Raman spectroscopy based on a single-crystal sapphire fiber. Opt. Lett. 36, 1287–1289 (2011). https://doi.org/10.1364/OL.36.001287
Levy, S., Alhadi, M.A., Azulay, A., Kahana, A., Bujanover, N., Gazit, R., McGargill, M.A., Friedman, L.M., Hertz, T.: FLU-LISA (fluorescence-linked immunosorbent assay): high-throughput antibody profiling using antigen microarrays. Immunol. Cell Biol. 101, 231–248 (2023). https://doi.org/10.1111/imcb.12618
Wu, Y., Guo, W., Peng, W., Zhao, Q., Piao, J., Zhang, B., Wu, X., Wang, H., Gong, X., Chang, J.: Enhanced fluorescence ELISA based on HAT triggering fluorescence “Turn-on” with enzyme-antibody dual labeled AuNP probe for ultrasensitive detection of AFP and HBsAg. ACS Appl. Mater. Interfaces 9, 9369–9377 (2017). https://doi.org/10.1021/acsami.6b16236
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant number 2020R1F1A1072912), the Korean government (MSIT) (Grant number 2018R1A5A2020732, 2019R1A2C2084122, and 2020R1A5A1018052), the Korea Institute for Advancement of Technology (KIAT) Grant funded by the Korean Government (MOTIE) (Grant numbers P0020967, P0017011, and 20000843), and the Ministry of Health & Welfare, Republic of Korea (Grant number HI22C1374).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (MP4 7802 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, SJ., Lee, K.H., Choi, YW. et al. Optical-Switch Raman Spectroscopy for High Throughput Screening. BioChip J 17, 318–328 (2023). https://doi.org/10.1007/s13206-023-00113-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13206-023-00113-3