
Abstract
A new expression cassette (EC0) consisting of the fused 5′ and 3′ intergenic regions (IGRs) of the Eremothecium cymbalariae translational elongation factor 1α (EcTEF1) gene was evaluated through expression of the bacterial hygromycin B phosphotransferase (hph) resistance gene in the common baker’s yeast Saccharomyces cerevisiae. Progressively shorter versions of the hph-containing EC cassette (hphEC1 though hphEC6) with trimmed 5′ and 3′ EcTEF1 IGRs were tested for their ability to confer resistance to hygromycin B in S. cerevisiae. Hygromycin B resistance was retained in all six generated hphEC variants up to a concentration of 400 mg/L. The hphEC6 cassette was the shortest cassette to be assayed in this study with 366 and 155 bp of the EcTEF1 5′ and 3′ IGRs, respectively. When tested for deletion of the S. cerevisiae proline oxidase gene PUT1, the hphEC6 cassette was shown to successfully act as a selection marker on hygromycin B-containing medium. The hphEC6 cassette could be placed immediately adjacent to a kanMX4 G418 disulfate resistance marker without any discernable effect on the ability of the yeast to grow in the presence of both hygromycin B and G418 disulfate. Co-cultivation experiments under non-selective conditions demonstrated that a PUT1 deletion strain carrying the hphEC6 cassette displayed equivalent fitness to an otherwise isogenic PUT1 deletion strain carrying the kanMX4 cassette.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the past 4 decades, the genetic engineering of yeasts and other microorganisms has become a multi-billion dollar industry. The common baker’s yeast Saccharomyces cerevisiae is the most commonly used yeast systems in synthetic biology applications although other species of budding yeasts (phylum Ascomycota, sub-phylum Saccharomycotina) are gaining in popularity. The traditional approaches to genetic engineering in yeast require condition-specific selection marker genes for the isolation of successful transformants. The design of dominant selection markers such as antibiotic resistance genes tends to employ promoter sequences from constitutively active and highly transcribed “house-keeping genes” such as those involved in protein synthesis, central carbon metabolism, or cell cycle progression. Strong constitutive promoters may also be desirable for some synthetic biology applications such as the rewiring of metabolic pathways or increasing pathway flux.
Translational elongation factor 1α (Tef1) is one of the most abundant proteins in actively dividing yeast cells (Ghaemmaghami et al. 2003) and the strong activity of the TEF1 gene promoter has lead to the development of a number of expression cassettes based on the TEF1 intergenic regions (IGRs) from yeasts (Müller et al. 1998; Ahn et al. 2007). The 5′ and 3′ IGRs of the TEF1 gene from the filamentous yeast Eremothecium gossypii (syn. Ashbya gossypii) has become one of the most widely used expression cassettes (commonly designated “MX”) for strong, constitutive expression of dominant selection markers in the genetic engineering of yeasts (Steiner and Philippsen 1994; Wach et al. 1994). The MX cassette has been shown to work for the expression of resistance genes in a wide spectrum of yeast species including the fission yeast Schizosaccharomyces pombe (Bähler et al. 1998) despite its very distant evolutionary relationship to budding yeasts (Beimforde et al. 2014).
However, the ubiquity of the MX expression cassette in the design of dominant selection markers for the genetic engineering of yeasts often results in transgenic strains that are modified several times with multiple MX cassettes carrying different resistance genes. The repeated use of the MX cassette or any other sequence element within genomic integration constructs will progressively decrease targeting efficiency of subsequent transformations within the same yeast genome (Davidson and Schiestl 2000). Accumulation of integrated sequence elements that contain longer stretches of identical sequence also introduces a risk of undesirable chromosomal rearrangements during genetic crosses and may introduce artifacts in high-throughput genetic analyses such as synthetic genetic arrays. Repeated use of a single or a limited sub-set of expression cassettes can also be problematic in synthetic biology applications such as heterologous expression of multigene biosynthetic pathways, where tandem arrays containing identical regulatory sequences can result in recombination-mediated excision of transgenes.
In the case of selection markers, one approach to circumvent the accumulation of identical regulatory elements within the genome of a yeast strain has been to remove the selection marker post-integration through heterologous expression of a recombinase such as Cre (Güldener et al. 1996). This approach has the added advantage of enabling repeated use of the same selection marker and thereby selection strategy. However, repeated recombinase-mediated selection marker recycling in S. cerevisiae has been shown to cause significant chromosomal rearrangements and gene loss (Solis-Escalante et al. 2015). An alternative approach is to introduce expression cassettes containing new promoter and terminator elements. For instance, there have been variants of the MX cassette described where the EgTEF1 terminator sequence have been substituted for that of other genes such as the S. cerevisiae ADH1 and CYC1 genes (Janke et al. 2004). Although this reduces the risk of unwanted recombination events with any pre-existing MX cassettes within the yeast genome, it also introduces a new risk of recombination events with the endogenous terminator elements of the ADH1 and CYC1 genes if these cassettes were to be used in the transformation of S. cerevisiae.
Newly sequenced yeast genomes are valuable resource for new regulatory sequences for heterologous expression of selection markers and other transgenes. As IGRs are non-coding, they evolve faster than coding gene sequences while retaining their regulatory function in related species. Thus, a promoter or terminator element can be transplanted into the genome of a more distantly related species and function, even though the overall sequence homology between promoters may be beyond detection. The present study sought to develop a new constitutive expression cassette based on the IGRs of the TEF1 gene from the filamentous yeast Eremothecium cymbalariae, which is the closest relative of E. gossypii with a sequenced genome at the time of writing (Wendland and Walther 2011).
Methods
Yeast integration constructs
PCR primers used in this study are listed in Table 1. An ScPUT1 targeting sequence (“Sc_Δput1”) consisting of two fused 200-bp segments of the ScPUT1 5′ and 3′ IGRs (GenBank accession AEHG01000269, residues 13,597–13,796 and 15,710–15,909, respectively) was synthesized de novo by GenScript (NJ, USA) and inserted into EcoRI/HindIII-cut pUC57 (GenBank accession Y14837) to produce the plasmid pUC57-Sc_Δput1 (Fig. 1a). The Sc_Δput1 sequence was amplified from pUC57-Sc_Δput1 using primers ScPUT1 3′ fwd and ScPUT1 5′ rev. The Sc_Δput1 amplification product was cut with BglII and SmaI and inserted in the reverse orientation into BglII/SmaI-cut pFA6a-kanMX4 (GenBank accession AJ002680; Wach et al. 1994) to produce the plasmid pFA6a-Sc_Δput1-kanMX4 (Fig. 1b, c).

Plasmid design. a Design of the S. cerevisiae PUT1 targeting cassette (Sc_Δput1) as inserted into the EcoRI/HindIII site of the pUC57 plasmid. b Sc_Δput1 targeting cassette was inserted into the pFA6a-kanMX4 plasmid in its reverse orientation. c Resulting pFA6a-Sc_Δput1-kanMX4 plasmid with insertion sites I and II highlighted in grey. Plasmid components are not drawn to scale. d Design of the E. cymbalariae TEF1 expression cassette (EC0) as inserted into the EcoRI/HindIII site of the pUC57 plasmid. The first four codons of the EcTEF1 ORF are highlighted in grey. e Schematic representation of the six hphEC cassettes assayed in this study. The lengths (bp) of 5′ and 3′ IGRs are indicated and drawn to scale. The hph coding sequence is not drawn to scale
A fusion of the full-length 5′ and 3′ IGRs of EcTEF1 gene (GenBank accession NC_016451, reverse complement of residues 841,757–842,469 and 840,021–840,379, respectively) was synthesized de novo by GenScript (NJ, USA) and inserted into EcoRI/HindIII-cut pUC57 to produce the plasmid pUC57-EC0 (Fig. 1d). The EC0 cassette also includes the first 12 basepairs of the EcTEF1 open reading frame (ORF) corresponding to the amino acid sequence MGKE. The hph coding sequence (GenBank accession K01193) was amplified from the plasmid pFA6a-hphNT1 (Janke et al. 2004) using primers hph fwd and hph rev. The hph amplification product was cut with Ecl136II and inserted into Ecl136II-cut pUC57-EC0 to produce the plasmid pUC57-hphEC0.
Six hphEC variants (hphEC1-hphEC6, Fig. 1e) were generated by PCR amplification of pUC57-hphEC0 using all six possible pair-wise combinations of forward primers EcTEF1 5′ 693 fwd, EcTEF1 5′ 581 fwd and EcTEF1 5′ 366 fwd with reverse primers EcTEF1 3′ 339 rev and EcTEF1 3′ 155 rev. Each hphEC variant (hphEC1 through hphEC6) was cut with MfeI and EcoRV and inserted into EcoRI/EcoRV-cut pFA6a-Sc_Δput1-kanMX4 (site I in Fig. 1c) to produce plasmids pFA6a-Sc_Δput1-kanMX4-hphEC1 through -hphEC6.
An “expression cassette-only” control plasmid (pFA6a-Sc_Δput1-kanMX4-EC1) was generated by first amplifying the 5′ and 3′ EcTEF1 IGRs from the pUC57-EC0 plasmid using primers EcTEF1 5′ 693 fwd and EcTEF1 3′ 339 rev. The resulting EC1 amplification product was then digested with MfeI and EcoRV and inserted into EcoRI/EcoRV-cut pFA6a-Sc_Δput1-kanMX4.
An integration plasmid where the kanMX4 marker had been replaced by the hphEC6 marker (pFA6a-Sc_Δput1-hphEC6) was generated by first excising the kanMX4 marker from the pFA6a-Sc_Δput1-kanMX4 plasmid through digestion with BglII and EcoRV. The hphEC6 cassette was amplified from the pUC57-hphEC0 plasmid with primers EcTEF1 5′ 366 fwd2 and EcTEF1 3′ 155 rev. The hphEC6 amplification product was then digested with BamHI and EcoRV and ligated to the BamHI/EcoRV-cut pFA6a-Sc_Δput1 fragment.
An alternative kanMX4/hphEC6 integration plasmid (pFA6a-hphEC6-Sc_Δput1-kanMX4) was generated by amplifying the hphEC6 cassette from the pUC57-hphEC0 plasmid with primers EcTEF1 5′ 366 fwd2 and EcTEF1 3′ 155 rev, which was then digested with BamHI and EcoRV and inserted into BamHI/SmaI-cut pFA6a-Sc_Δput1-kanMX4 (site II in Fig. 1c).
Prior to transformation, integration plasmids were digested with SwaI to produce linearized integration constructs (Fig. 2a), which were then purified into sterile water using the QIAquick PCR purification kit (Qiagen).

Integration of plasmid constructs at the S. cerevisiae PUT1 locus. a pFA6a-Sc_Δput1-kanMX4 plasmid was linearized by digestion with SwaI to enable homologous recombination with PUT1 5′ and 3′ IGRs. The locations of control primers to confirm correct integration of the construct are indicated. DNA elements are not drawn to scale. b Confirmation of the correct integration of constructs as demonstrated by PCR of genomic DNA using primers pFA6a fwd and ScPUT1 5′ ctrl rev. c Confirmation of the removal of the endogenous PUT1 locus as demonstrated by PCR of genomic DNA using primers ScPUT1 3′ fwd and ScPUT1 ctrl rev
Yeast transformation
The transgenic yeast strains generated in this study are listed in Table 2. The parent yeast strain for all transformations in this study was S. cerevisiae CBS 8340 (syn. CEN.PK 113-7D; genotype MATa MAL2-8c SUC2), which was purchased from Centraalbureau voor Schimmelcultures (Utrecht, The Netherlands). The selection agents G418 disulfate and hygromycin B were purchased from Formedium Ltd (Norfolk, UK). An aqueous stock solution of hygromycin B was prepared to a final concentration of 50 g/L, sterilized by filtration and stored at 4 °C. An aqueous stock solution of G418 disulfate was prepared to a final concentration of 100 g/L, sterilized by filtration, and stored as aliquots at − 20 °C.
The transformation protocol used in this study is a simplified version of the standard lithium acetate protocol. A S. cerevisiae pre-culture was diluted to a final OD600 of 0.1 in 50-mL fresh YM broth (3-g/L yeast extract, 3-g/L malt extract, 5-g/L peptone, and 10-g/L glucose) supplemented with 75-mg/L carbenicillin and incubated at 30 °C with shaking at 200 rpm until OD600 reached 0.5–0.6. Cells were collected by centrifugation (2500×g, 5 min) and resuspended in 1.5-mL sterile water. The washed cells were collected by centrifugation (6900×g, 3 min), the supernatant removed and the pellet resuspended in 0.4-mL 100-mM lithium acetate. The resuspended cells were incubated at 30 °C for 15 min and then divided into 50-μL aliquots. 10-μL purified SwaI-digested integration plasmid was added to each aliquot of cells, which were then incubated at 30 °C for 15 min. 0.3 mL of a 100 mM lithium acetate/40% (w/v) PEG3350 mixture was added to each sample, and mixed and incubated for a further 15 min at 30 °C. Samples were then incubated for 15 min at 42 °C followed by immediate centrifugation (6900×g, 3 min) and removal of the supernatant. 0.5 mL of fresh YM broth was added to each sample, which was left to stand for 5 min at room temperature before being fully resuspended. Each sample of 0.5-mL resuspended cells were transferred to a fresh tube containing 2.5-mL fresh YM broth and incubated at 30 °C for 1 h in a rotary shaker set to 200 rpm. Samples were then centrifuged (6900×g, 3 min) and the supernatant discarded. The cells in each sample were resuspended in 0.2-mL fresh YM broth and spread on solid YM medium (20 g/L agar) containing either 400-mg/L G418 disulfate or 200-mg/L hygromycin B as specified in the “Results and discussion” section.
Correct chromosomal integration and the deletion of the PUT1 locus were confirmed by PCR analysis of purified genomic DNA from each strain (Fig. 2a). Successful integration at the PUT1 locus was assayed using primers pFA6a ctrl fwd and ScPUT1 5′ ctrl rev, which produce no product in the CBS 8340 parent strain, a 417-bp amplification product in strains TLSC001–TLSC009, and a 1980-bp amplification product TLSC010 due to the insertion of the hphEC6 cassette at site II (Fig. 2b). Successful deletion of the PUT1 coding sequence was assayed using primers ScPUT1 3′ ctrl fwd and ScPUT1 ctrl rev, which produce a 1063-bp amplification product in the CBS 8340 parent strain and no product in strains TLSC001–TLSC010 (Fig. 2c).
Comparative fitness assays of deletion strains
The absolute fitness of G418 disulfate/hygromycin B-resistant strains TLSC008 and TLSC010 was compared by parallel cultivation in minimal glucose medium (MMD) broth consisting of 6.7 g/L Difco yeast nitrogen base without amino acids (Becton, Dickinson, and Company) and 20 g/L glucose. Both strains were pre-cultured in 3 mL MMD with 200 mg/L G418 disulfate and 200 mg/L hygromycin B. Pre-cultures were diluted to a final OD600 of 0.05 in separate 250-mL E-flasks containing 40-mL MMD broth with 200-mg/L G418 disulfate and 200-mg/L hygromycin B. Samples were incubated at 30 °C with shaking at 200 rpm and growth was monitored by measurement of OD600 every 24 h for 4 days. Each strain was assayed in triplicate.
The relative fitness of G418 disulfate-resistant strain TLSC001 and hygromycin B-resistant strain TLSC009 were assayed through co-cultivation of both strains in MMD broth without selection agent. Both strains were pre-cultured in 3-mL MMD containing either 200-mg/L G418 disulfate (for strain TLSC001) or 200-mg/L hygromycin B (for strain TLSC009). 1 mL of each pre-culture was harvested by centrifugation (6900×g, 3 min) and resuspended in an equal volume of fresh MMD broth without selection agent. Both strains were inoculated to a final OD600 of 0.01 each in a single 250-mL E-flask containing 40-mL MMD broth without selection agent and incubated at 30 °C with shaking at 200 rpm. Growth of each strain was monitored through enumeration by viable count on YM solid medium containing either 200 mg/L G418 disulfate (for strain TLSC001) or 200 mg/L hygromycin B (for strain TLSC009). Co-cultures were sampled immediately following inoculation as well as after 24 and 48 h after inoculation. Co-culture experiments were performed in triplicate. Viable counts at each time point was performed in triplicate and the average expressed as colony forming units (cfu)/mL.
Results and discussion
The abundance of sequenced yeast genomes is a valuable resource for co-opting intergenic regulatory sequences for heterologous expression of transgenes in yeast. In this study, the promoter and terminator elements contained within the 5′ and 3′ IGRs of the E. cymbalariae TEF1 gene (EcTEF1, systematic gene name Ecym_3450) were selected for the development of a new constitutive expression cassette in yeast. At the time writing, the yeast E. cymbalariae is the closest relative of E. gossypii with a sequenced genome (Wendland and Walther 2011). The EcTEF1 5′ and 3′ IGRs were, therefore, considered logical choices for the development of a new expression cassette considering the relatively short evolutionary distance to the well established EgTEF1-based MX cassette.
A fusion of the full-length 5′ and 3′ EcTEF1 IGRs was synthesized de novo and inserted into the pUC57 cloning vector. The EcTEF1 expression cassette (from now on referred to as EC0) also included the first four codons of the EcTEF1 ORF corresponding to the amino acids MGKE (Fig. 1d). This differs slightly from the EgTEF1-derived MX cassette, which includes the first eight codons of the EgTEF1 ORF corresponding to the amino acids MGKEKTHV. The rationale for including a shorter N-terminal portion of the E. cymbalariae Tef1 peptide sequence was to reduce the possibility of interference with the N-terminus of transgenes that could be critical to the function of the expressed protein. The EcTEF1 5′ and 3′ IGRs are both significantly longer than those of the E. gossypii ortholog. The EcTEF1 5′ IGR consists of 713 bp compared to 284 bp for the EgTEF1 5′ IGR (GenBank accession NC_005785, reverse complement of residues 58,674–58,957), while the EcTEF1 3′ IGR consists of 359 bp compared to 165 bp in the EgTEF1 3′ IGR (GenBank accession NC_005785, reverse complement of residues 57,132–57,296).
The 5′ and 3′ IGRs from E. cymbalariae and E. gossypii TEF1 genes were aligned to assess their degree of sequence conservation. The majority of the EgTEF1 5′ IGR sequence appeared to be conserved in the EcTEF1 5′ IGR, while the EcTEF1 5′ IGR contained two longer stretches of sequence that were not conserved in the EgTEF1 (Fig. S1a). The largest conserved sequence block (318 bp with 53% identity and six short indels comprising a total of 57 internal gap positions) between the EcTEF1 and EgTEF1 5′ IGRs was located immediately upstream of the TEF1 ORF in both species. The majority of EgTEF1 3′ IGR appeared to be conserved in the EcTEF1 3′ IGR with the majority of the conserved sequence contained within a single 156-bp sequence block (64% identity with a single internal 8-bp indel), which was located immediately downstream of the TEF1 ORF in both species.
Both TEF1 5′ IGRs were inspected for the presence of known conserved regulatory elements that are commonly found in the promoter sequences of ascomycete ribosomal protein (RP) genes (Tanay et al. 2005). Of previously characterized RP gene regulatory elements, a single putative Homolo-D site (TGTGACTG) as well as an immediately adjacent putative Rap1-binding site (CRCCCRTACAT) were found to be conserved in the 5′ end of both TEF1 5′ IGRs (Fig. S1a). A putative ribosomal RNA processing element (RRPE) motif (AAAAATTTT) was detected in the E. cymbalariae TEF1 5′ IGR, which was not conserved in the E. gossypii TEF1 5′ IGR (Fig. S1a). The 6-bp core sequence AATTTT of the RRPE motif is also associated with the regulation of genes required for rapid growth in budding yeasts (Ihmels et al. 2005) and is referred to as the rapid growth element (RGE). A putative RGE motif was detected in the E. cymbalariae TEF1 5′ IGR located just upstream of the putative RRPE/RGE motif. This RGE site was not conserved in the E. gossypii TEF1 5′ IGR either. Further inspection of the full-length E. gossypii TEF1 5′ IGR sequence identified a single putative RGE motif in the reverse orientation. However, this site overlapped with a conserved region in the E. cymbalariae TEF1 5′ IGR sequence, which may indicate that it is not a de facto regulatory element.
It is possible that the 318-bp conserved sequence block just immediately upstream of the TEF1 ORF in both species may contain novel regulatory elements important for the proper expression of the TEF1 gene. The genus Eremothecium belongs to the family Saccharomycetaceae, which includes several species with sequenced genomes such as S. cerevisiae, lactose-fermenting biotechnology yeasts of the genus Kluyveromyces, and osmotolerant spoilage yeasts of the genus Zygosaccharomyces among others. The abundance of available genomic sequences from species belonging to the Saccharomycetaceae enables the mining of genomic data for the presence of novel conserved motifs in the TEF1 gene. However, the TEF1 gene is found in two genomic contexts within the Saccharomycetaceae. In species belonging to the genera Eremothecium, Kluyveromyces, Lachancea, Torulaspora, and Zygosaccharomyces, the TEF1 gene is arranged in a tandem orientation with its neighboring upstream and downstream genes MRL1 and MUD1, respectively (Fig. S1b). Conversely, the TEF1 gene is present in two near-identical paralogous copies (TEF1 and TEF2) in the genera Kazachstania, Naumovozyma, Saccharomyces, Tetrapisispora, and Vanderwaltozyma. These genera constitute a monophyletic group originating from an ancient whole-genome duplication (WGD) event (Kellis et al. 2004). The WGD group TEF1 paralog has retained the ancestral MRL1-TEF1 IGR, while the TEF2 paralog has retained the ancestral TEF1-MUD1 IGR (Fig. S1c).
To scan for conserved sequence motifs in the TEF1 5′ IGR among the Saccharomycetaceae, the ancestral MRL1-TEF1 IGR (with the MRL1 and TEF1 genes in a tandem orientation) must first be distinguished from the more recently derived TKL2-TEF2 IGR found within the WGD group (with the TKL2 and TEF2 genes in a divergent orientation). In the latter case, the TKL2 and TEF2 genes share a common bidirectional promoter, which could introduce unwanted noise into the motif elucidation algorithm. Consequently, all TKL2-TEF2 IGR sequences from WGD species of the Saccharomycetaceae were excluded from the further analysis in the current study. MRL1-TEF1 IGR sequences from 24 species belonging to the Saccharomycetaceae (Eremothecium coryli, E. cymbalariae, E. gossypii, Kazachstania africana, Kazachstania naganishii, Kluyveromyces aestuarii, Kluyveromyces lactis, Kluyveromyces marxianus, Kluyveromyces wickerhamii, Lachancea kluyveri, Lachancea lanzarotensis, Lachancea quebecensis, Lachancea thermotolerans, Lachancea waltii, Naumovozyma castellii, Naumovozyma dairenensis, S. cerevisiae, Saccharomyces eubayanus, Saccharomyces paradoxus, Tetrapisispora blattae, Tetrapisispora phaffii, Torulaspora delbrueckii, Zygosaccharomyces bailii, and Zygosaccharomyces rouxii) were assayed for the overrepresentation of sequence motifs using MEME (http://meme-suite.org/tools/meme; Bailey et al. 2009). The only significant hits were the previously identified RRPE motif, which was detected in 19 out the 24 input sequences, and the shorter RGE motif, which was found in all 24 sequences. Putative Homolo-D and Rap1 sites were only found in a smaller sub-set of the sequences. No novel sites above the significance threshold (E = 10−3) were identified.
The greater lengths of the 5′ and 3′ IGRs of the EcTEF1 gene as compared to those of the EgTEF1 gene may be problematic for some applications: for example, decreased PCR amplification efficiency of longer linear DNA constructs. Such problems could be circumvented by trimming one or both IGRs of the EcTEF1 gene as long as sufficient functionality could be retained. To test the potential of using trimmed variants of the EC0 cassette, the plasmid-encoded hygromycin B phosphotransferase gene hph from the bacterium Escherichia coli was selected for expression. Hygromycin B is an aminoglycoside antibiotic produced by the actinobacterium Streptomyces hygroscopicus (Mann and Bromer 1958), which inhibits protein synthesis in both bacteria and eukaryotes. Expression of the hph gene has previously been shown to confer resistance to hygromycin B in both bacteria and yeast (Gritz and Davies 1983; Kaster et al. 1984).
The hph coding sequence was inserted into the EC0 cassette to generate the hphEC0 cassette. A spectrum of hphEC variants (hphEC1 through hphEC6) with progressively trimmed 5′ and 3′ IGR segments was subsequently generated by PCR amplification using different primer combinations (Fig. 1e, Fig. S1a). Each hphEC variant was then inserted into the integration construct pFA6a-Sc_Δput1-kanMX4 (Fig. 1c), so that the hphEC cassette was located immediately downstream of the kanMX4 selection marker in a tandem orientation. The S. cerevisiae proline oxidase-encoding PUT1 locus was selected as the integration site for this study as the gene is only essential for cell viability when l-proline is the only available source of nitrogen (Wang and Brandriss 1986), which allows for convenient phenotypical confirmation of transformants. A control construct containing an empty EC1 cassette (pFA6a-Sc_Δput1-kanMX4-EC1) was included to confirm that the EcTEF1 IGRs did not confer cryptic resistance to hygromycin B.
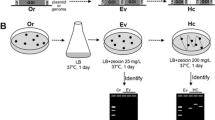
The integration constructs were used to transform S. cerevisiae with G418 disulfate used for selection of positive transformants. Plate growth assays of confirmed transformants containing any of the six hphEC variants (strains TLSC003-TLSC008) were able to grow on solid YM medium containing up to 400 mg/L hygromycin B, which was the highest concentration tested (Fig. 3). No significant growth was observed on hygromycin B-containing medium (100–400 mg/L) for strains TLSC001 and TLSC002, which had been transformed with the control constructs pFA6a-Sc_Δput1-kanMX4 and pFA6a-Sc_Δput1-kanMX4-EC1, respectively.

Hygromycin B tolerance of hphEC variants. Strains were pre-cultured overnight in 3 mL YM broth with 200 mg/L G418 disulfate and then diluted to OD600 0.1 in fresh YM broth without any selection agent. 2 μL cell suspension of each strain was spotted on solid YM medium with the indicated concentration of hygromycin B. Plates were incubated for 2 days at 30 °C and then photographed
The previously observed putative Homolo-D and Rap1 sites in the EcTEF1 5′ IGR (Fig. S1a) were shown to be dispensable for sufficient expression of the hph gene in S. cerevisiae to enable growth at hygromycin B concentrations up to 400 mg/L as demonstrated by hphEC variants hphEC3 through hphEC6. This does not exclude the possibility that these two motifs do play a role in the proper regulation of the endogenous TEF1 gene in E. cymbalariae. Another possibility is that the MRL1 gene, which is located immediately upstream of TEF1 in the E. cymbalariae genome (Fig. S1b), is the regulatory target of these sites. However, the MRL1 gene encodes a putative membrane protein similar to mammalian mannose-6-phosphate receptors with a possible function as a sorting receptor in the delivery of vacuolar hydrolases (Whyte and Munro 2001) but does not appear to have any obvious functional connection to protein synthesis. In addition, the E. cymbalariae MRL1 ortholog (systematic gene name Ecym_3451) is annotated as non-functional with a reported frame-shift mutation. The tandem orientation of the MRL1 and TEF1 genes would also suggest that the conserved Homolo-D and Rap1-binding sites in the MRL1-TEF1 IGR, if functional, are not involved in regulating the expression of the MRL1 gene.
All remaining experiments of the hphEC cassette within the current study utilized the shortest variant, hphEC6. The next question to be addressed was whether the adjacent kanMX4 and hphEC6 cassettes in the pFA6a-Sc_Δput1-kanMX4-hphEC6 construct interacted in any positive manner or negative manner. For example, would the hphEC6 selection marker retain its activity in the absence of the kanMX4 cassette? Likewise, would the close proximity of the kanMX4 and hphEC6 cassettes in the pFA6a-Sc_Δput1-kanMX4-hphEC6 construct cause any detectable interference between the two selection markers?
Two additional hphEC6-containing constructs were, therefore, generated to investigate any potential interaction between the hphEC6 and kanMX4 cassettes in the pFA6a-Sc_Δput1-kanMX4-hphEC6 construct. The first construct replaced the kanMX4 cassette in the pFA6a-Sc_Δput1-kanMX4 plasmid with the hphEC6 cassette to produce the plasmid pFA6a-Sc_Δput1-hphEC6 (Fig. 4a). This construct would enable the evaluation of the hphEC6 marker both as a selection marker for transformation on hygromycin B-containing medium and also allow functional study of the hphEC6 cassette in the absence of the kanMX4 cassette. The transformation of S. cerevisiae CBS 8340 was repeated with the pFA6a-Sc_Δput1-hphEC6 construct with 200 mg/L hygromycin B as the selection agent rather than 400 mg/L G418 disulfate. Ten random colonies were picked from the selective media and screened for the presence of the pFA6a-Sc_Δput1-hphEC6 construct at the PUT1 locus by PCR as before (Fig. 2b, c). All ten transformants were found to have successfully integrated the pFA6a-Sc_Δput1-hphEC6 construct and replaced the endogenous PUT1 locus, which demonstrated the utility of the hphEC6 as a hygromycin B-dependent selection marker. One of the transformants was selected for subsequent assays and designated strain TLSC009.

Assaying interactions and relative fitness between kanMX4 and hphEC6 selection markers. a Outline of linearized constructs used in interaction assays. Plasmid elements are not drawn to scale. b Tolerance of yeast strains to combinations of G418 sulfate and hygromycin B. Strains were pre-cultured overnight in 3 mL YM broth with either 200 mg/L G418 disulfate (TLSC001), 200 mg/L hygromycin B (TLSC009), or 200 mg/L of both selection agents (TLSC008 and TLSC010). Pre-cultures were diluted to OD600 0.1 in fresh YM broth without any selection agent. 2 μL cell suspension of each strain was spotted on solid YM medium with the indicated concentration of G418 disulfate and hygromycin B. Plates were incubated for 2 days at 30 °C and then photographed. c Comparative growth dynamics in MMD broth containing both G418 disulfate and hygromycin B. Error bars indicate one standard deviation. d Fitness equivalence of co-cultivated S. cerevisiae Δput1 strains carrying either the kanMX4 or hphEC6 marker under non-selective conditions. Error bars indicate one standard deviation
A second construct was designed where the hphEC6 cassette was inserted at site II in pFA6a-Sc_Δput1-kanMX4 (Fig. 1c) to make plasmid pFA6a-hphEC6-Sc_Δput1-kanMX4 (Fig. 4a). Once the linearized pFA6a-hphEC6-Sc_Δput1-kanMX4 construct had been integrated into the S. cerevisiae genome to make strain TLSC010, the kanMX4 and hphEC6 cassettes would be separated by the entire length of the pFA6a plasmid backbone (2479 bp). The tolerance of strains TLSC009 and TLSC010 to G418 disulfate and hygromycin B were tested using a plate growth assay and compared to strains TLSC001 and TLSC008 (Fig. 4b). As expected, strain TLSC009 was sensitive to G418 at 200 mg/L, while strain TLSC010 was resistant to both G418 disulfate and hygromycin B at 200 mg/L.
Although strains TLSC008 and TLSC010 displayed similar growth on solid medium containing both G418 disulfate and hygromycin B, the agar growth assay was only considered semi-quantitative. A growth assay in liquid medium was, therefore, performed to investigate whether the proximity between the kanMX4 and hphEC6 cassettes in strains TLSC008 and TLSC010 had any subtle effect of strain fitness when both resistance markers are under selection. Both strains were cultivated in parallel using a chemically defined liquid minimal medium containing both G418 disulfate and hygromycin B. The growth dynamics of both strains were monitored by optical density measurements every 24 h and shown to be both highly reproducible and indistinguishable from each other (Fig. 4c). It was, therefore, concluded that the hphEC6 cassette could be placed immediately adjacent to the kanMX4 cassette in a tandem orientation without any discernable effects. However, the kanMX4 and hphEC6 cassettes were not tested in a convergent orientation in this study and there is a possibility that the selection markers could affect the activity of each other in such an orientation.
The final question to be resolved in the present study was whether the hphEC6 cassette was functionally equivalent to the kanMX4 cassette in terms of strain fitness under non-selective conditions where neither selection agent is present. Relative fitness was assayed through co-cultivation of strains TLSC001 and TLSC009, which are isogenic with exception of carrying either the kanMX4 or hphEC6 cassette, respectively (Fig. 4a). Both strains were inoculated in chemically defined liquid minimal medium lacking any selection agent with an initial OD600 of 0.01 corresponding to 65,000–70,000 cfu/mL of each strain. Growth of both strains was monitored separately through enumeration by viable count on solid-rich medium containing either G418 disulfate or hygromycin B, respectively. Both strains had reached stationary phase within 24 h and no significant deviation from the initial TLSC001/TLSC009 inoculation ratio (1.04 ± 0.16) was observed at either 24 or 48 h after inoculation (Fig. 4d). It could not be excluded that significant differences in fitness between the selection markers would be observed after prolonged incubation in stationary phase. Likewise, such differences in fitness may become apparent only after repeated cycles of batch co-cultivations. However, for standard genetic applications such as heterologous expression of dominant selection markers to enable gene replacement, the kanMX4 and hphEC6 selection markers appear to be essentially equal in fitness.
Conclusion
The present study has demonstrated the suitability of the EcTEF1-derived expression cassette EC0 for the design of dominant selection markers in yeast engineering. The EcTEF1 5′ and 3′ IGRs could be trimmed down to at least 366 and 155 bp, respectively, while retaining resistance to at least 400 mg/L hygromycin B when expressing the hph hygromycin phosphotransferase gene (Fig. 3). Detailed expression dynamics of different variants of the EC cassette with regard to growth phase and nutritional status were not investigated in this study. Such information may be of importance when employing the cassette for the expression of transgenes for other purposes than dominant selection markers and would need to be determined on a case-by-case basis both in terms of transgene and strain background. Concerning the latter aspect, the differences in RP gene regulatory elements in different ascomycete fungi (Tanay et al. 2005) need to be considered as they may affect expression dynamics in a species-dependent manner.
Abbreviations
- EcTEF1 :
-
Eremothecium cymbalariae translational factor 1α gene
- hph :
-
Hygromycin phosphotransferase gene
- IGR:
-
Intergenic region
- ORF:
-
Open reading frame
- RGE:
-
Rapid growth element
- RP:
-
Ribosomal protein
- RRPE:
-
Ribosomal RNA processing element
- ScPUT1 :
-
Saccharomyces cerevisiae proline oxidase gene
References
Ahn J, Hong J, Lee H, Park M, Lee E, Kim C, Choi E, Jung J, Lee H (2007) Translation elongation factor 1-alpha gene from Pichia pastoris: molecular cloning, sequence, and use of its promoter. Appl Microbiol Biotechnol 74:601–608
Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14:943–951
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren JY, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208
Beimforde C, Feldberg K, Nylinder S, Rikkinen J, Tuovila H, Dörfelt H, Gube M, Jackson DJ, Reitner J, Seyfullah LJ, Schmidt AR (2014) Estimating the Phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol Phylogenet Evol 78:386–398
Davidson JF, Schiestl RH (2000) Mis-targeting of multiple gene disruption constructs containing hisG. Curr Genet 38:188–190
Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425:737–741
Gritz L, Davies J (1983) Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene 25:179–188
Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24:2519–2524
Ihmels J, Bergmann S, Gerami-Nejad M, Yanai I, McClellan M, Berman J, Barkai N (2005) Rewiring of the yeast transcriptional network through the evolution of motif usage. Science 309:938–940
Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21:947–962
Kaster KR, Burgett SG, Ingolia TD (1984) Hygromycin B resistance as dominant selectable marker in yeast. Curr Genet 8:353–358
Kellis M, Birren BW, Lander ES (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428:617–624
Mann RL, Bromer WW (1958) The isolation of a second antibiotic from Streptomyces hygroscopicus. J Am Chem Soc 80:2714–2716
Müller S, Thomas S, Peter KH, Henrik D (1998) Comparison of expression systems in the yeast Saccharomyces cerevisiae, Hansenula polymorpha, Kluyveromyces lactis, Schizosaccharomyces pombe and Yarrowia lipolytica. Cloning of two novel promoters from Yarrowia lipolytica. Yeast 14:1267–1283
Solis-Escalante D, van den Broek M, Kuijpers NG, Pronk JT, Boles E, Daran JM, Daran-Lapujade P (2015) The genome sequence of the popular hexose-transport-deficient Saccharomyces cerevisiae strain EBYVW4000 reveals LoxP/Cre-induced translocations and gene loss. FEMS Yeast Res 15 pii:fou 004
Steiner S, Philippsen P (1994) Sequence and promoter analysis of the highly expressed TEF gene of the filamentous fungus Ashbya gossypii. Mol Gen Genet 242:263–271
Tanay A, Regev A, Shamir R (2005) Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci USA 102:7203–7208
Wach A, Brachat A, Pohlmann R, Philippsen P (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808
Wang SS, Brandriss MC (1986) Proline utilization in Saccharomyces cerevisiae: analysis of the cloned PUT1 gene. Mol Cell Biol 6:2638–2645
Wendland J, Walther A (2011) Genome evolution in the eremothecium clade of the Saccharomyces complex revealed by comparative genomics. G3 1:539–548
Whyte JR, Munro S (2001) A yeast homolog of the mammalian mannose 6-phosphate receptors contributes to the sorting of vacuolar hydrolases. Curr Biol 11:1074–1078
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Conflict of interest
The author declares no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Analysis of the TEF1 IGRs in E. cymbalariae and E. gossypii. a The EcTEF1 and EgTEF1 5′ and 3′ IGRs were aligned in MAFFT (https://mafft.cbrc.jp/alignment/server/). Identical sequence positions are shaded maroon. Sequence positions of the trimmed EcTEF1 5′ and 3 ‘IGR variants assayed in this study are indicated with arrows. Putative Homolo-D, Rap1, RRPE and RGE sites are highlighted. The EcTEF1 coding sequence is indicated by the orange block arrow and is not drawn to scale. b Genomic context of the TEF1 locus among non-WGD genera of the Saccharomycetaceae (Eremothecium, Kluyveromyces, Lachancea, Torulaspora, Zygosaccharomyces). c Genomic context of the TEF1 and TEF2 loci among WGD genera of the Saccharomycetaceae (Kazachstania, Naumovozyma, Saccharomyces, Tetrapisispora, Vanderwaltozyma). The direction of arrows indicate whether each ORF is located on the forward or reverse strand with respect to the TEF1/TEF2 ORF. ORFs and IGRs are not drawn to scale. (EPS 621 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Linder, T. Development of a yeast heterologous expression cassette based on the promoter and terminator elements of the Eremothecium cymbalariae translational elongation factor 1α (EcTEF1) gene. 3 Biotech 8, 203 (2018). https://doi.org/10.1007/s13205-018-1224-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-018-1224-0