
Abstract
A novel nickel-based methanation catalyst has been prepared and tested for CO and CO2 hydrogenation to synthetic natural gas under various conditions. The catalysts before and after reaction have been characterized using XRD, Laser Raman and IR spectroscopes. It is showed that the novel catalyst can give high methane selectivity and yield at a very broad ranges of temperature and H2/CO ratios, although the selectivity to methane increase with the H2/CO ratio, which, however, increases with the temperature rising from 200 to 350 °C, then gradually decrease. The characterization results showed that the there is surface carbons forming over the catalyst surface, and the nickel crystalline structure changes during the reaction. Despite this, the catalyst still gives the same performance, which suggested that the catalyst can operate in a very broad range conditions.
Similar content being viewed by others

Introduction
Biomass development is predicted to grow the fastest of all renewable energy in the next decade, although it is already being utilized for a variety of purposes, including biofuels, biopower (electricity), biomaterials, biochemicals (“green chemistry”), and biopellets. The different categories of biomass are all interrelated, and of all the sources of renewable energy, biomass can be most honestly labeled “home-grown.” Among these applications, Synthetic Natural Gas (SNG) production from biomass has attracted increasing attention in recent years, due to the rising price of natural gas, the wish for less dependency from natural gas imports and the opportunity of reducing green house gases. Although solid dry biomass has [1–5] been used directly (e.g., wood and straw) for ages, converting them into SNG has is a more preferential process.
The advantages SNG are, besides the relatively high efficiency of its production, the already existed infrastructure such as pipelines and the efficient end use technologies such as compressed natural gas cars, combined heat and power plants, or combined cycle plant. Therefore, in recent years there has been a dramatic increase interest in the manufacture of a clean, high BTU gas energy source which will meet pipeline standards by synthetic means biomass.
SNG normally is produced through the gasified products, e.g., H2 and CO-containing gas stream. While a number of metallic species are known to be active and selective methanation catalysts including, inter alia, nickel, ruthenium, cobalt and iron, their application to the manufacture of high BTU or pipeline gas has been less than satisfactory for several reasons. First, methanation reactions are strong exothermic reaction, which easily heats up catalyst to 800 °C in an adiabatic reactor. However, these catalyst systems generally cannot stand very high temperature, the reactor temperature needs to be limited to temperatures below 400 °C to avoid sintering and deactivation of the catalyst, so the highly exothermic nature of the methanation reaction itself provides severe operational difficulties in controlling catalyst temperature when CO concentration of the feed gas is in the range required for methane-rich gas manufacture. Further, the methanation reaction itself is considered to be a combination of several reactions including the primary reaction and secondary reactions (2) and (3)
The thermodynamic calculation results for this process are shown in Table 1.
These thermodynamic equilibriums of the reaction 1 shows that the equilibrium yield of methane is adversely effected at high temperatures, i.e., above 900 K, as can be seen, the delta G at 900 K is +1,742 J/mol, which does not favor the methanation reaction. In fact, it can be seen that reaction (2) is a combination of reaction (1) and the water gas shift reaction (4).
So far, there have been 3 processes proposed for the SNG production, one is high temperature processes [6–10], which requires very highly stable catalyst and several steps for heat exchange, so as to cool down the reaction stream to improve the catalyst performance and avoid the catalyst deactivation. Another is to the low-temperature methanation [11–13], which proceeds in a reaction configuring with heat exchanger so as to remove the reaction heat quickly, and may have low requirements on the catalyst stability, but requires the catalyst to have high activity and selectivity. So far, the methanation catalyst is normally nickel-based system, which is prepared using impregnation method. The catalyst thus has high dispersion which can also be used in fluidized bed reaction and slurry reactor for SNG production, where the heat transfer is very fast, but attrition is a more important parameter [1, 14–18], also the separation of the catalyst particles from the reactor is also an issue.
Recently, a novel reactor system has been developed by Boxenergy Tech Ltd, which can take the reaction heat away in a fixed bed reactor, thus to maintain a constant low isothermal reaction temperature. In such case, the catalyst with low temperature activity and selectivity is more important. Overview of the previous publications, there are few reports on the low-temperature methanation catalyst for SNG production. It has shown that the organic matrix combustion method can give a Fischer–Tropsch catalyst with super high activity [19, 20], thus in this work, a novel method, e.g., organics assisted catalyst preparation method has been developed, and tested under various conditions, a very nickel based robust methanation catalyst with high activity and selectivity has been developed.
Experimental
Catalyst preparation
The catalyst has been prepared using organic induced partial combustion method. It may be an evolution of the chelating and organic combustion method [21, 22]. In short, a specific amount of 20.8 g Ni(NO3)2·6H2O, 1.06 g ZrO(NO3)2, and 5 g citric acid are mixed with 20 ml of water, heating to 50 °C while stirring for 30 min to form a transparent solution. 10 g of AlOOH (beomite) is then added to the solution, and statically placed under ambient conditions for 4 h, dried at 80 °C for 2 h and calcined in static air at 500 °C for 4 h to give a black powder. The nickel content in the black powder is analyzed using atom absorption, and the content is 30.1 %. The resultant powder is pelletized using a mechanical pelletizing machine. When using for catalyst, the sample is crushed and sieved into 60–100 mesh particles.
Catalyst testing
Every time, 0.1 g of the crushed NiZrO x /Al2O3 catalyst is loaded in a quartz tube as the reactor. The reactor is inserted into a tubular furnace and connected to the gases. The catalyst is activated with 33.3 (vol) % H2/N2 flowing at 70 ml/min and heated at 2 °C/min to 350 °C and held at this temperature for 2 h. Then the reactor is cooled down to 200 °C in the flowing reduction atmosphere.
The gas is switched to a gas mixture of N2:H2:CO = 60:30:10 (volume) with a total following rate of 100 ml/min. when testing CO2 hydrogenation, the mixture gas is set as N2:H2:CO2 = 50:40:10. The other settings are all the same. The products flow through a cold trap where the produced vapour is condensed the flowing rate of the gas in and out were measured using a soap bubble meter, and the dried gaseous product concentration are analyzed using non-dispersive IR spectrometer.
The CO conversion and methane selectivity and yield are calculated as follows:
where VCO, in and out are the volume flowing rate under the ambient conditions calculated from the flowing rate and the content of CO and CH4 in the inlet and outlet gas stream.
Catalyst characterization
XRD diffraction was carried out in a Philips PW1710 diffractometer equipped with Cu Kα radiation to detect the crystalline phase of nickel and alumina and coke after reaction. The morphology of the catalysts before and after reaction was observed in a JEOL-4000EX high-resolution electron microscope with an accelerating voltage of 400 kV. Raman spectra were recorded with a resolution of 2 cm−1 using a Yvon Jobin Labram spectrometer with an He+ laser, running on a back scattered confocal arrangement.
Results and discussion
Normally when CO is hydrogenated, it is firstly converted into –CH2– which either link together to form C n H m as liquid hydrocarbons. This is a well-known process for Fischer–Tropsch synthesis [23–26]. For SNG production, –CH2– is expected to further hydrogenated into CH4 rather than link to form the liquid. In such case, C2+ selectivity is more important. In SNG production, CH4 selectivity as well as yield is more important [13, 15, 27, 28]. Therefore in the following test results, only CH4 selectivity and yield are presented under various conditions.
Figure 1 gives the temperature effect on the CH4 selectivity and yield of CO hydrogenation test under different conditions. It is seen that the main product is methane, no C2+ hydrocarbons were detected, the methanation reaction starts up from 200 °C, however, significant CH4 yield appear from 220 °C, which may be due to the low CO conversion, although the methane selectivity is significantly high. The CO selectivity reaches 87 % at 220 °C and almost unchanged from 220 °C to the reaction temperature to 400 °C, while the yield of methane decreases gradually with the methanation temperature. This can be explained by the strong exothermicity of the methanation reaction. The higher methanation temperature may lead to some steam reforming reaction, which is strong endothermic reaction. It is, therefore, inferred that the methanation reaction should be kept at temperatures from 260 to 300 °C. However, as showed before, once methanation starts, the generated heat is huge, thus how to remove the reaction heat away quickly is an issue when applying the low-temperature methanation process [29].

Effect of temperature on the methanation reaction. Test conditions: GHSV = 60,000 h−1, N2:H2:CO = 6:3:1
In contrast to the CO methanation, CO2 hydrogenation to methane showed different trends. As shown in Fig. 2, the CO2 hydrogenation starts up at 200 °C, although the methane yield is significantly lower, only about 1.4 % over the NiZrO x /Al2O3 catalyst. When the reaction temperature is raised to 250 °C, methane selectivity increases to 95 %, while the CH4 yield is about 35 %. When the methanation temperature rises to 280 °C, methane selectivity reaches about 98 %, while the CH4 yield is about 62 %, suggesting that the catalyst prepared in this way gave a high CH4 selectivity, although its activity for CO2 conversion is lower than that for CO hydrogenation. The CH4 yield reaches the maximum at 340 °C, while the CH4 selectivity is about 98 %, suggesting that the suitable operation conditions for CO2 methanation is 340 °C when pressure is 1 bar. Also the methanation temperatures above 340 °C have less effect on the CH4 selectivity, and CO2 conversion cannot reach more than 90 %.

Effect of reaction temperature on CO2 methanation under the conditions of: GHSV = 60,000 h−1, P: 1 bar; N2:H2:CO2 = 5:4:1
CO2 methanation performance with the temperature can be explained by the reaction process steps. Generally, CO2 hydrogenation has two steps
Reaction 5 is endothermic, and reaction 6 is strong exothermic. The overall reaction heat is less than CO methanation, therefore its CH4 yield increase in a broader temperature range than the CO only methanation.
The stability of the catalyst for CO methanation with time at 250 °C and 1 bar and GHSV of 60,000 h−1 is shown in Fig. 3. The CO conversion under this condition at the start of run is nearly 100 %, although the selectivity to methane is about 95 %, and the CH4 yield is 92.2 %. The CH4 selectivity drops to nearly 94 % after 3.5 h running, but changes to 95 %, which drops again after 8 h to 94 %. This change might result from the analysis errors, because the gas products were detected by non-dispersive IR. Thus, it can be inferred that the CH4 selectivity in fact does not change much during the 600 min time on stream. However, the CH4 yield as shown by the blue curve in Fig. 3 changes from 92.1 % at the start of the reaction to 89.8 %, which suggests that the CO conversion may drops during the reaction. From the decline trend it can be found that the yield drops faster in the first 260 min, but slower after 430 min. This can be explained by that the fresh catalyst in the initial reaction stage may be more sensitive to the reactants, the reactant can induce the catalyst surface change or leads to carbon deposition, which may cover some active site, thus leading to the activity drop. With the time on stream, the active site tends to go to steady state, the carbon deposition rate is less or equivalent to the carbon hydrogenation rate, thus the yield tends to be stable. It is interesting to see that although the CH4 yield drops slowly with the time on stream, which may be due to the loss of some active site or the nickel metal sintering, the methane selectivity over the supported nickel catalyst almost unchanged, suggesting that the catalyst active site change or nickel metal structure change does not alter the methanation reaction on the other active sites.

Stability test of CO methanation for SNG production under the conditions of: Methanation reaction conditions: H2:CO:N2 = 30:10:60, P: 1 bar; reaction temperature: 250 °C, GHSV: 60,000 h−1
The effect of H2/CO ratio on the products selectivity has also been studied over the NiZrO x /Al2O3 catalyst. A methanation process normally involves in CO absorption over nickel site, which is then attacked by H2 to give CH4 and H2O [30]. If there is not enough H2 around the activated CO, it may convert into carbon and CO2. So when H2/CO ratio is 1.5, the selectivity to methane is about 80 %, and CO2 about 20 %, no other hydrocarbons are produced. This suggests that the prepared nickel catalyst can work under low H2/CO ratio gas mixture. With the H2/CO ratios increasing, the CO2 selectivity gradually drops, while CH4 selectivity increases, this is in agreement with the thermodynamic prediction (Fig. 4). When H2/CO ratio reaches the theoretical value for methanation, the CH4 selectivity is about 84 %, while there is still some CO2 generated, which may be due to competitive reaction between the Boudart reaction and methanation [23, 31].

Effcet of H2/CO ratio on CO methanation. The test conditions are shown in the graph
Overview of the above test results shows that the NiZrO x /Al2O3 catalyst prepared in this way has high methane selectivity, little C2+ side products are generated. The catalyst showed high activity at low temperature and can operate in a wide range.
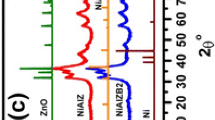
The catalyst before and after catalytic test have been characterized using XRD, and the results are shown in Fig. 5. For XRD pattern (the green curve in Fig. 5) of the as prepared catalyst only showed three broad diffraction peaks, which are assigned to the diffraction of gama-Al2O3 [32–34]. When the catalyst is tested for CO methanation in the mixture of [(N2 + CO2)/H2 = 4] at 350 °C for 5 h in stream, while the catalyst still had CH4 selectivity more than 90 % and CO conversion more than 89 %, the XRD patterns of the alumina became sharper, compared to the fresh catalyst sample. This suggests that the catalyst may experience partly crystallization during the reaction. Also diffraction peaks at 38.7, 43.6, 58.5 and 62.5 appeared in this used sample. According to the literature, the peaks at 38.7 and 58.5 are due to the metallic nickel crystal with hcp structure, while the diffraction peaks at 43.6 and 62.5 are ascribed to the diffraction of metallic nickel with fcc structure. The peak of fcc form nickel is much stronger than that of hcp, suggesting that the main phase of nickel after CO2 hydrogenation is fcc structure [35].

XRD patterns of the methanation catalysts at various stages
XRD patterns of the spent catalyst from the CO methanation reaction for 600 min is different from the catalyst unloaded from CO2 methanation reaction. Probably the CO methanation reaction was controlled at 250 °C, the Al2O3 support diffraction peak is not as sharp as that from CO2 system, suggesting that Al2O3 support still keeps most of its amorphous phases. Besides these peaks, there are small diffraction bands at 43.8°, 52.8° and 63.2° observed, which correspond to the diffraction of planes of 011, 200 and 220 of nickel crystal. Meanwhile, a small sharp diffraction peak at 27.6° is seen, which can be assigned to the deposited carbon [36, 37]. This suggests that there is carbon formed during the CO hydrogenation although the catalyst is still active.
The spent catalysts from different methanation reactions under various conditions have been measured using Laser Raman spectroscopy. In the fresh catalyst, almost no Raman band can be seen. The Raman spectrum of the spent catalyst after CO2 methanation showed two distinct peaks at 1,404 and 1,882 cm−1, and a shoulder peak at around 1,590 cm−1. According to literatures, the Raman band at 1404 cm−1 can be assigned to the D band, and that at 1,590 cm−1 is due to the G band [38, 39]. The D band has arisen from structure defects or imperfection of graphite, whereas the G band is associated with a splitting of the E2g stretching mode of graphite. In addition, a very weak D′ band is present at ca. 1,600 cm−1 as a shoulder of the G band, which has stemmed from the dangling band of disorder graphite. The broad bang at 1,882 cm−1 is unknown, and may need further study.
In the Raman spectrum of the spent catalyst from CO methanation, these peaks intensity increases significantly, suggesting that both D and G bands graphite increase significantly. This implies that over the spent catalyst from CO methanation more carbon is formed over the catalyst surface, which is in agreement with the XRD results (Fig. 6).

Laser Raman spectra of the methanation catalyst at different stages
The TEM images of the nickel catalyst from CO methanation and CO2 methanation are shown in Fig. 7. The image of the spent catalyst from CO2 has smaller particles, the nickel particles size ranges from 20 to 60 nm. No whisker carbon is observed in the catalyst surface. The dark black fake may results from nickel particle or amorphous carbon as suggested by Laser Raman. However, in the TEM images of spent nickel from CO methanation, clearly the nickel particles and the surface carbon sizes are bigger, as suggested by XRD and Laser Raman. More carbon formed in the spent catalyst from CO methanation, However, no whisker carbon were seen over the catalyst, which might be the reason the catalyst is still active even after 600 min time on stream, because it is already pointed out that the whisker carbon normally account for the catalyst deactivation [36, 37].

TEM of the spent NiZrO x /Al2O3 catalysts from a CO2 methanation at 300 °C; b CO methanation at 250 °C
Conclusion
Al2O3 supported Ni catalyst for CO x methanation to produce synthetic natural gas has been prepared using organic decomposition method and tested under various conditions.
The catalyst prepared from this method catalyses CO methanation from 220 °C and CO2 methanation starting from 260 °C. It showed high CH4 selectivity and yield, the catalyst can operate in a broad range of temperature.
The catalyst showed high stability and CH4 selectivity for CO methanation, although more surface coke is detected over the catalyst surface.
The nickel crystallite becomes bigger and has different forms in the spent catalyst from CO2 methanation reactions, while the one from CO methanation has different metallic form and more crystalline carbon.
References
van der Meijden CM et al (2008) Production of Bio-CNG by gasification. In: Proceedings of 25th Annual International Pittsburgh Coal Conference, pp 133/1–133/8
Weiss AJ (1973) CRG [catalytic rich gas]-hydrogasification process for SNG [synthetic natural gas] production. Chem Eng Progr 69(5):84–90
Woodward C (1977) Methanation in substitute natural gas production. Energiespectrum 1(12):342–347
Wix C (2010) Process for the production of substitute natural gas. (Den.). Application, US, p 10
Lessard RR, Reitz RA (1981) Catalytic coal gasification: an emerging technology for SNG. Baytown Res. Dev. Div., Exxon Res. Eng. Co., Baytown, TX, USA, p 26
Hoehlein B et al (1984) Methane from synthesis gas and operation of high-temperature methanation. Nucl Eng Des 78(2):241–250
Luterbacher Jeremy S et al (2009) Hydrothermal gasification of waste biomass: process design and life cycle assessment. Environ Sci Technol 43(5):1578–1583
Rostrup-Nielsen JR, Pedersen K, Sehested J (2007) High temperature methanation. Appl Catal A 330:134–138
Udengaard NR, Olsen AN, Wix-Nielsen C (2006) High temperature methanation process-revisited. In: Proceedings of 25th annual international Pittsburgh coal conference, pp 25/1–25/5
Woodward C (1976) A high-temperature methanation catalyst for SNG applications. Am Chem Soc Div Fuel Chem Prep 21(4):22–29
Shinnar R, Fortuna G, Shapira D (1982) Thermodynamic and kinetic constraints of catalytic synthetic natural gas processes. Ind Eng Chem Process Des Dev 21(4):728–750
Yang Z et al (2010) Isothermal methanation for manufacture of substitute natural gas from coal or biomass. (Shanghai International Engineering Consulting Company, Peop. Rep. China). Application, CN, p 18
Kopyscinski J, Schildhauer TJ, Biollaz SMA (2010) Production of synthetic natural gas (SNG) from coal and dry biomass—a technology review from 1950 to 2009. Fuel 89(8):1763–1783
Bock HJ et al (1986) The Comflux-process for substitute natural gas (SNG). In: Proceedings of Eng. Found. Conf. Fluid, pp 489–496
Seemann MC, Schildhauer TJ, Biollaz SMA (2010) Fluidized bed methanation of wood-derived producer gas for the production of synthetic natural gas. Ind Eng Chem Res 49(15):7034–7038
van der Meijden CM et al (2009) Bioenergy II: scale-up of the MILENA biomass gasification process. Int J Chem React Eng 7
Blinn MB et al (1989) Advanced gasifier-desulfurizer process development for SNG (substitute natural gas) application. Final report August 1987–December 1988. KRW Energy Syst., Inc., Pittsburgh, PA, USA, p 210
Punwani DV, Arora JL, Tsaros CL (1978) SNG from peat by the PEATGAS Process. Inst. Gas Technol., Chicago, IL, USA, p 19
Xiao T, Chen H (2011) Methanation catalyst, its preparation process, and methanation reaction device having the same, Peop. Rep. China, p 11
Xiao T, Qian Y (2008) Promoted carbide-based Fischer-Tropsch catalyst, method for its preparation and uses thereof. Oxford Catalysts Limited, UK, p 35
Rezaei M et al (2006) Nanocrystalline zirconia as support for nickel catalyst in methane reforming with CO2. Energy Fuels 20(3):923–929
Seo JG et al (2008) Preparation of Ni/Al2O3–ZrO2 catalysts and their application to hydrogen production by steam reforming of LNG: effect of ZrO2 content grafted on Al2O3. Catal Today 138(3–4):130–134
Boudart M, McDonald MA (1984) Structure sensitivity of hydrocarbon synthesis from carbon monoxide and hydrogen. J Phys Chem 88(11):2185–2195
Dalai AK, Davis BH (2008) Fischer-Tropsch synthesis: a review of water effects on the performances of unsupported and supported Co catalysts. Appl Catal A 348(1):1–15
Khodakov AY (2009) Fischer-Tropsch synthesis: relations between structure of cobalt catalysts and their catalytic performance. Catal Today 144(3–4):251–257
Stiegel GJ, Srivastava RD (1994) Natural gas conversion technologies. Chem Ind (London) 21:854–856
Bery RN (1973) SNG [substitute natural gas] from naphtha. Chem Eng (N. Y.) 80(13):90–91
Woodcock KE, Hill VL (1987) Coal gasification for synthetic natural gas (SNG) production. Energy (Oxford) 12(8–9):663–687
Tao P, Wang X (2007) Method for preparing synthetic natural gas (SNG) from coke oven gas (COG). (Southwest Research and Design Institute of Chemical Industry, Peop. Rep. China). Application, CN, p 9
Vogel F et al (2007) Synthetic natural gas from biomass by catalytic conversion in supercritical water. Green Chem 9(6):616–619
Ichikawa S, Poppa H, Boudart M (1985) Disproportionation of carbon monoxide on small particles of silica-supported palladium. J Catal 91(1):1–10
Hao Z et al (2009) Characterization of aerogel Ni/Al2O3 catalysts and investigation on their stability for CH4-CO2 reforming in a fluidized bed. Fuel Process Technol 90(1):113–121
Jun J et al (2008) Surface chemistry and catalytic activity of Ni/Al2O3 irradiated with high-energy electron beam. Appl Surf Sci 254(15):4557–4564
Xiao T-C et al (2004) Tungsten promoted Ni/Al2O3 catalysts for carbon dioxide reforming of methane to synthesis gas. Chem Res Chin Univ 20(4):470–477
Li H et al (2008) Fabrication and growth mechanism of Ni-filled carbon nanotubes by the catalytic method. J Alloys Compd 465(1–2):51–55
Koo KY et al (2008) Coke study on MgO-promoted Ni/Al2O3 catalyst in combined H2O and CO2 reforming of methane for gas to liquid (GTL) process. Appl Catal A 340(2):183–190
Martinez R et al (2004) CO2 reforming of methane over coprecipitated Ni–Al catalysts modified with lanthanum. Appl Catal A 274(1–2):139–149
Reshetenko TV et al (2003) Catalytic filamentous carbon. Structural and textural properties. Carbon 41(8):1605–1615
Zhu X, Cheng D, Kuai P (2008) Catalytic decomposition of methane over Ni/Al2O3 catalysts: effect of plasma treatment on carbon formation. Energy Fuels 22(3):1480–1484
Acknowledgments
This work has been financially supported by Guangzhou Bairen (Hundred Talents) program. We would like to thank Dr. Zhenxing Liang for his discussion and professor Jixin Su for his activity test.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Huang, Y., Chen, H., Su, J. et al. Highly active and selective catalyst for synthetic natural gas (SNG) production. Appl Petrochem Res 4, 181–188 (2014). https://doi.org/10.1007/s13203-013-0034-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-013-0034-x