
Abstract
Background
Various components of the immunological milieu surrounding tumors have become a key focus in cancer immunotherapy research. There are currently no reliable biomarkers for triple-negative breast cancer (TNBC), leading to limited clinical benefits. However, some studies have indicated that patients with TNBC may achieve better outcomes after immunotherapy. Therefore, this study aimed to identify molecular features potentially associated with conventional type 1 dendritic cell (cDC1) immunity to provide new insights into TNBC prognostication and immunotherapy decision-making.
Methods
Single-cell ribonucleic acid sequencing data from the Gene Expression Omnibus database were analyzed to determine which genes are differentially expressed genes (DEGs) in cDC1s. We then cross-referenced cDC1-related DEGs with gene sets linked to immunity from the ImmPort and InnateDB databases to screen for the genes linked to the immune response and cDC1s. We used univariate Cox and least absolute shrinkage and selection operator regression analyses to construct a risk assessment model based on four genes in patients with TNBC obtained from the Cancer Genome Atlas, which was validated in a testing group. This model was also used to assess immunotherapy responses among the IMvigor210 cohort. We subsequently utilized single sample Gene Set Enrichment Analysis, CIBERSORT, and ESTIMATE to analyze the immunological characteristics of the feature genes and their correlation with drug response.
Results
We identified 93 DEGs related to the immune response and cDC1s, of which four (IDO1, HLA-DOB, CTSD, and IL3RA) were substantially linked to the overall survival rate of TNBC patients. The risk assessment model based on these genes stratified patients into high- and low-risk groups. Low-risk patients exhibited enriched ‘‘hot tumor’’ phenotypes, including higher infiltration of memory-activated CD4 + T cells, CD8 + T cells, gamma delta T cells, and M1 macrophages, as well as elevated immune checkpoint expression and tumor mutational burden, suggesting potential responsiveness to immunotherapy. Conversely, high-risk patients displayed “cold tumor” characteristics, with higher infiltration of M0 and M2 macrophages and lower immune scores, which may be poorer in response to immunotherapy. However, experimental validation and larger clinical studies are necessary to confirm these findings and explore the underlying mechanisms of the identified genes.
Conclusion
This study developed a robust risk assessment model using four genes that effectively forecast the outcome of patients with TNBC and have the potential to guide immunotherapy. This model provided new theoretical insights for knowing the TNBC immune microenvironment and developing personalized treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Triple-negative breast cancer (TNBC) is characterized by a lack of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2 (also known as the lack of fluorescence in situ hybridization amplification of human epidermal growth factor receptor 2), and accounts for approximately 15–20% of all breast cancer cases [1]. TNBC has a worse prognosis than other subtypes of breast cancer because of its extremely aggressive clinical course, high rates of metastasis and recurrence, low survival rates, and few available therapeutic choices [2, 3]. While chemotherapy is still the most common treatment for TNBC, tumor responses are often short-lived, resulting in unsatisfactory long-term outcomes. Studies have shown, however, that immunotherapy may offer improved clinical benefits in TNBC patients.
The advent of immunotherapy has altered the way that many malignant cancers are treated, especially immune checkpoint inhibitors (ICIs) [4]. TNBC is more likely to benefit from immunotherapy than other subtypes of breast cancer because of several significant features. First, the high mutational burden and genomic instability of TNBC can cause additional tumor-specific neoantigens to be produced [5]. Second, TNBC is typically rich in lymphocytes that infiltrate tumors and are linked to better prognoses in early stages of the disease [6]. Lastly, elevated expression of programmed death-ligand 1 (PD-L1) in TNBC provides a direct target for ICI therapy [7]. Given the immunogenic characteristics of TNBC, targeting ICIs for the treatment of TNBC, such as adjuvant chemotherapy combined with PD-1/PD-L1, shows significant promise [8, 9]. Most patients, however, do not achieve sustained clinical responses to ICIs and response rates to treatment remain limited [10]. Therefore, it is crucial to explore immune biomarkers and therapeutic targets that predict treatment outcomes to enhance the efficacy of immunotherapy in patients with TNBC.
In the tumor microenvironment (TME), the initiation and regulation of both innate and adaptive responses depend on dendritic cells (DCs), which are important antigen-presenting cells. Conventional type 1 dendritic cells (cDC1s) are a critical subpopulation of DCs essential for anticancer immunity [11]. Several investigations using preclinical mouse cancer models have shown that cDC1s are essential for T cell-mediated immunogenic tumor rejection and the response to ICIs [12,13,14]. An abundance of cDC1s in human malignancies is linked to better clinical responses to ICIs and survival rates for cancer patients [15, 16]. cDC1s' crucial function in anti-cancer immunity is partly reflected in its capacity to absorb and transport tumor cell antigens to lymph nodes that drain tumors, thereby eliciting tumor-specific cytotoxic CD8 + T cells [12, 17, 18]. Furthermore, in addition to triggering T-cell responses, recent studies have shown that cDC1s play a critical role in directly regulating cytotoxic T-cell immunity within the TME. By presenting tumor antigens and producing cytokines that alter T cell survival and effector function, intratumoral cDC1s can generate chemokines to draw CD8 + T cells into the TME and reactivate and multiply anti-cancer CD8 + T cells [19,20,21]. However, our current understanding of the clinical significance and regulatory mechanisms of cDC1s in TNBC is incomplete. Therefore, a deeper knowledge of the genes linked to immunity controlling cDC1s may reveal new targets for immunotherapy in TNBC.
Using single-cell ribonucleic acid sequencing (scRNA-seq), we analyzed 16 TNBC samples and 3 normal breast tissue samples from 19 patients. This study sought to create an extensive single-cell atlas of TNBC. We thoroughly examined this atlas to learn about the various kinds of classical immune cells in the TNBC microenvironment. Furthermore, we combined a significant amount of RNA sequencing data with scRNA-seq data from patients with TNBC and were successful in identifying a group of genes linked to cDC1s that are immune-related. We subsequently developed a robust and reliable four-gene signature (IDO1, HLA-DOB, CTSD, and IL3RA) prognostic risk model using the least absolute shrinkage and selection operator (LASSO) and univariate Cox analysis, which is capable of precisely determining the risk levels of TNBC patients. We systematically assessed the differences between the groups at high and low risk according to the final risk classification. Additionally, we created a brand-new nomogram by fusing risk scores and clinical characteristics. Finally, to identify variations in immunological characteristics, medication responses, and expression patterns, we used CIBERSORT, ESTIMATE, as well as single sample Gene Set Enrichment Analysis (ssGSEA) to contrast the TNBC groups at high and low risk. These discoveries not only improved our knowledge of the TNBC microenvironment but also offered encouraging opportunities to improve prognosis and diagnosis in the clinical context of TNBC.
2 Methods
2.1 Data collection
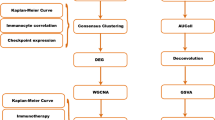
This work made use of three datasets from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/): GSE199515-GPL24676, GSE176078-GPL18573 [22], and GSE161529-GPL18573 [23]. We selected research samples from these three datasets and ultimately identified 19 patient samples, including TNBC (n = 16) and normal breast (NORMAL; n = 3) tissues. Samples were selected based on the following inclusion criteria: (1) The TNBC status or normal breast tissue was clearly annotated. (2) High-quality scRNA-seq data were available. (3) Patient demographic and clinical information were provided when possible. Conversely, samples were excluded if they met the following criteria: (1) Low-quality sequencing data were exhibited. (2) The TNBC status or normal breast tissue had incomplete or unclear annotations. Additionally, prognostic model training data for patients with TNBC, including gene expression matrices, clinical details, and mutation details, were taken from the Cancer Genome Atlas (TCGA) at https://portal.gdc.cancer.gov/. The GEO database was also used to extract the cohort for external validation, which comprised gene expression and clinical data (GSE58812-GPL570) and was used to evaluate the predicted accuracy of our model. Local ethics committee approval was not required because the data were obtained from public databases (GEO and TCGA). The publication standards and data access policies issued by TCGA and GEO were closely followed in this investigation [24, 25]. Figure 1 illustrates the overall workflow, including data collection, preprocessing, clustering, differentially expressed genes (DEGs) identification and model validation, as well as the prognostic analysis and immunotherapy potential evaluation of triple-negative breast cancer based on the traditional type 1 dendritic cell immune-related gene prediction model.

Workflow for prognostic analysis and evaluation of immunotherapy potential based on conventional type 1 dendritic cell (cDC1) immune-related genes in triple-negative breast cancer (TNBC)
2.2 Data processing for scRNA-seq
We processed the scRNA-seq data to ensure its quality and suitability for downstream analysis. Initially, each sample's barcodes, features, and matrix data were retrieved and utilized to create objects in Scanpy [26]. The following steps were performed for data filtering and normalization: (1) Low-quality cells were excluded by removing cells expressing fewer than 200 genes and with genes expressed in fewer than 3 cells; (2) potential doublets were identified and excluded using the Scrublet tool; (3) cells with more than 6,000 expressed genes and those with mitochondrial gene counts exceeding 25% of the total were filtered out to ensure data quality; (4) the library size of each cell was normalized using Scanpy's scanpy.pp.normalize_total function to account for technical variability; (5) a log-normalized data matrix was generated to prepare the data for downstream analysis. This rigorous step-by-step approach ensured reproducibility and accuracy in scRNA-seq data preprocessing.
2.3 Dimensionality reduction and unsupervised clustering
To perform decrease of dimensionality and unsupervised clustering, we executed a series of processes based on the Scanpy workflow. First, we used the scanpy.pp.highly_variable_genes function to select highly variable genes, selecting the top 4,000 for further examination. The impact of total counts and the proportion of mitochondrial gene expression for each cell were then regressed out using the scanpy.pp.regress_out technique. Next, we utilized the “max_value = 10” option when using the scanpy.pp.scale method to scale the expression values of each gene to unit variance. After preprocessing the data, we reduced the dimensionality through principal component analysis. To eliminate batch effects between the different datasets, we ensured data integration and consistency using the sc.external.pp.harmony_integrate function with the parameter “n_pcs = 30.” Finally, we utilized the scanpy.tl.tsne function to implement the t-SNE algorithm, which stands for t-distributed stochastic neighbor embedding, further reducing the dimensionality of the merged dataset for visualization and analysis of cell distribution. For the clustering analysis, we utilized the Leiden clustering method to cluster the cell neighborhood graph and identify the cell populations.
2.4 Cell cluster annotation
We utilized experimentally validated marker genes computed using Scanpy to accurately annotate the cell clusters. These marker genes are highly specific and are expressed within cell clusters. First, we identified the cell types associated with marker genes using Panglao DB (https://panglaodb.se/) and CellMarker (http://biocc.hrbmu.edu.cn/CellMarker/) [27]. The CellTypist tool (https://github.com/Teichlab/celltypist) [28] was subsequently employed to determine the cell-type annotations for every cluster based on the known marker gene expression patterns.
2.5 Acquisition of immune-related DEGs
In order to determine DEGs for cDC1s, we first used the Wilcoxon rank-sum test to compare each gene's expression in a particular cluster to that in other cells. Significant DEGs were defined as genes that satisfied the following requirements: Initially, almost 25% of the target cluster's cells expressed the genes; second, the genes showed log2(fold change) values > 1 or < -1, indicating considerable overexpression within the intended cluster; finally, for the differential expression analysis to be statistically significant, the adjusted P-value needs to be less than 0.05. We used R's ggplot2 program to construct a volcano plot that showed the variations in these genes' expression. The ImmPort (https://www.immport.org/home) and InnateDB (https://www.innatedb.com/) databases provided us with 2,533 genes associated with immunity. Immune-related DEGs were identified by intersecting cluster-specific DEGs with genes curated in ImmPort and InnateDB.
2.6 Pathway enrichment analysis of Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG)
We used the R package clusterProfiler to perform KEGG pathway and GO functional analyses in order to gain a better understanding of the roles of immune-related DEGs in cDC1s [29, 30]. In the GO analysis, specifically, we concentrated on molecular functions, cellular components, and biological processes to comprehensively assess the functional characteristics of the genes. Additionally, a KEGG pathway study was used to comprehend the participation of these genes in specific biological pathways. The criterion for statistical significance is established at an adjusted P-value of less than 0.05 in order to guarantee the data's statistical significance.
2.7 Building and verifying a prognostic model
We first used a univariate Cox analysis to find predictively significant cDC1 immune-related genes in order to develop a consistent and dependable prognostic model for patients with TNBC. We then utilized LASSO regression to further filter these prognostic cDC1 immune-related genes and build the prognostic model. This model made it possible to use the following method to determine a risk score for every patient with TNBC.
Where Expi indicates the messenger RNA expression level of the i-th gene and Coefi represents its regression coefficient in LASSO-Cox. Patients from the TCGA-TNBC dataset and external validation cohort (GEO dataset GSE58812) were categorized into groups based on the median rating, at high and low risk. The survival and survminer R packages were utilized to display the results, and Kaplan–Meier survival curves were employed to evaluate the survival outcomes. We also generated receiver operating characteristic (ROC) curves and calculated the area under the curve (AUC) using the timeROC R package to validate the prognostic efficacy of the derived risk scores, thereby quantifying the predictive accuracy of the risk model. Lastly, we validated the independent prognostic variables using univariate and multivariate Cox regression analysis as well as ROC curve validation.
2.8 Nomogram construction
To further assess the predictive power of the risk score model, we constructed a nomogram using the rms R package, incorporating other clinicopathological characteristics of patients with TNBC including age, stage, and TNM classification [31]. The concordance index (C-index) was calculated to assess the nomogram's predictive accuracy and gauge the degree of agreement between the predicted and actual survival rates. The x-axis represented the anticipated odds of survival for each patient at 1, 3, and 5 years, whereas the y-axis indicated the actual survival probabilities, with a 45° line representing the optimal prediction. In order to evaluate the nomogram’s net clinical benefit, we also looked at the decision curve.
2.9 Tumor mutation analysis and chemotherapy response assessment
To evaluate tumor mutation burden (TMB), we utilized the maftools package in R [32], followed by the pRRophetic framework to evaluate five popular chemotherapeutic medications' half-maximal inhibitory concentrations (IC50s) (docetaxel, paclitaxel, gemcitabine, doxorubicin, and cisplatin) [33]. In brief, using the largest pharmacogenomic database available to the public, the Genomics of Drug Sensitivity in Cancer database (https://www.cancerrxgene.org/), we forecasted each sample’s response to chemotherapy. Ridge regression was used to determine the IC50 estimations, and all parameters were left at their default settings.
2.10 Immune assessment analysis
We initially estimated the immune infiltration scores and relative proportions of 22 immune cell types from the TCGA-TNBC database using CIBERSORT for the immune assessment study of the high- and low-risk groups. CIBERSORT uses support vector machine techniques to connect each gene expression profile with a reference cell type spectrum. Then, using the GSVA R package and ssGSEA, we conducted enrichment analysis of 29 immune characteristics. Next, we used the ESTIMATE R program to predict immune and stromal cell scores based on gene expression patterns. Since these molecules are frequently targeted in ICI therapies. Next, to evaluate the effectiveness of anti-PD-L1 immunotherapy, we used the IMvigor210 cohort to examine the expression of CD274 (PD-L1), CTLA4, LAG3, and PDCD1 (PD1) in high- and low-risk patients.
2.11 Statistical analyses
Statistical analyses were performed using R (v4.1.0) and Python (v3.8) to ensure robust and reproducible results. Covariate adjustments were implemented using multivariable Cox proportional hazards regression, accounting for potential confounders such as age, tumor stage, and histological grade. Model performance metrics, including C-index, calibration curves, and decision curve analyses, were computed to evaluate the prognostic model. Data visualization and statistical validation were conducted with the ggplot2 and survminer packages in R.
3 Results
3.1 scRNA-seq atlas construction for TNBC
We examined 19 patients using scRNA-seq to ascertain the cellular makeup of the TME in TNBC by using 16 TNBC and 3 normal breast tissue samples.We meticulously assessed for annotated typical cell markers and batch effects (as mentioned above), enabling us to classify the 86,021 cells into 11 main cell lineages: DCs, fibroblasts, endothelial cells, pericytes, plasma cells, macrophages, B cells, natural killer (NK) cells, ductal epithelial cells, and CD4 and CD8 + T cells lymphocytes (Fig. 2A). These major lineages each expressed specific classical marker genes (Fig. 2B), confirming the accuracy of the classification of our lineage. Figure 2C illustrates the consistency of the cellular makeup of different patients, whereas Fig. 2D provides an overview of single-cell data from normal tissue and tumor samples. Of note, Fig. 2E highlights the differing proportions of the 11 aforementioned cell types in normal vs. tumor tissues, exhibiting a larger number of several immune cell types in the tissues of the tumor. This observation may be linked to enhanced immune infiltration in the TNBC samples. Given the significant variation in DC populations among patients and their crucial roles in immune responses, we further categorized DCs into four major subpopulations using CellTypist (Fig. 2F). Interestingly, the proportions of these four major DC subpopulations (Fig. 2G) varied across tissues, with cDC1s, cDC2s, and pDCs showing increased proportions in TNBC tissues, whereas migratory DCs were more prevalent in normal tissues.

Comprehensive single-cell atlas of TNBC: A major cell lineages from samples of 19 patients; B bubble plot of classical marker gene expression in every major cell lineage; C employing t-distributed stochastic neighbor embedding (t-SNE) to analyze samples from 19 patients; D t-SNE analysis of normal and tumor samples; E proportions and counts of every type of cell in samples of tumors and normal cells; F t-SNE analysis of dendritic cells (DCs) classified by cell type; G proportions and counts of DC subpopulations in normal and tumor samples
3.2 GO functional analysis of four subpopulations of DCs
To explore the functional differences among the four DC subpopulations, we conducted a GO analysis on the genes of each subpopulation (Fig. 3). The analysis's findings showed that in regard to biological processes, cDC1s and cDC2s primarily participate in immune responses, whereas pDCs are closely associated with the regulation of cytokines, such as the positive regulation of cytokine production. In contrast, the functions of migratory DCs are closely associated with viral responses, exemplified by their response to viruses. The primary roles of class I molecules of the major histocompatibility complex (MHC) are played by cDC1s, including MHC class I receptor activity, whereas cDC2s are connected to MHC class II substances, such as MHC class II protein complexes' interaction. Previous studies have shown that MHC class I molecules are essential for presenting intracellular antigens and making them recognizable to cytotoxic CD8 + T cells. Decrease in MHC class I molecules may allow cancer cells to avoid T cell-mediated cytotoxicity adaptive immune responses. Furthermore, MHC class I molecules absence in TNBC may present possible obstacles to the effectiveness of PD-1/PD-L1 inhibitors [34]. Therefore, a deeper understanding of the genes that regulate cDC1s may offer fresh targets for the disease's treatment.

Gene Ontology (GO) functional analysis of the four DC subpopulations: A functional analysis of cDC1 genes in GO; B functional analysis of cDC2 genes in GO; C functional analysis of pDC genes in GO; D functional analysis of migratory DC genes in GO
3.3 Construction of cDC1 immune-related DEGs
By comparing the gene profiles of cDC1s with those of other DC subtypes, we filtered out 361 DEGs (|Log2(FC)| > 1, adjusted P < 0.05), which were visualized as a volcano plot (Fig. 4A). We subsequently performed additional filtering by matching the 361 DEGs with 2,533 genes linked to immunity from the ImmPort and InnateDB databases, leading to a finished set of 93 DEGs associated with immunity and cDC1s (Fig. 4B, Supplementary Table S1). To clarify the potential biological mechanisms underlying these cDC1 immune-related DEGs, we used the KEGG and GO databases for our research. The molecular mechanisms primarily responsible for leukocyte-mediated immunity, adaptive immunological responses based on immune cell somatic recombination, lymphocyte-mediated immunity, and immune effector process regulation were revealed by our analysis. Molecular functions included immune receptor activity, antigen binding, immunoglobulin binding, and MHC protein binding (Fig. 4C, Supplementary Table S2). According to KEGG analysis, these DEGs were primarily associated with primary immunodeficiency, natural killer cell-mediated cytotoxicity, antigen processing and presentation, and the B-cell receptor signaling pathway (Fig. 4D, Supplementary Table S3).

Construction of cDC1 immune-related differentially expressed genes (DEGs): A DEGs in the volcano plot comparing cDC1 with other DC subtypes, with red representing upregulated genes and blue representing downregulated genes; B Venn diagram showing genes linked to immunity and DEGs associated with cDC1s; C potential biological mechanisms of DEGs linked to the immune system in cDC1s, emphasizing the variety of molecular functions, cellular constituents, and biological processes found in GO analysis; D identification of important pathways based on examination of the Kyoto Encyclopedia of Genes and Genomes
3.4 Prognostic model construction based on cDC1 immune-related genes
In order to ascertain the prognosis of TNBC, we employed a univariate Cox proportional hazards regression analysis to ascertain the connection between the 93 immune-related DEGs identified in cDC1s. Among the 93 genes, 4 genes showed a robust association with the TCGA cohort's general survival (P < 0.05) (Fig. 5A). Following the identification of prognostic genes, LASSO regression was utilized to build a strategy for predicting risk based on these four genes linked to survival, through which they were identified as robust prognostic indicators for TNBC (Figs. 5B, C). To assess the patient risk, we constructed a strategy for predicting risk based on these four genes linked to survival and calculated using the formula to determine the risk score for the coefficient: risk rating = (− 0.173 × IDO1) + (− 0.595 × HLA-DOB) + (0.678 × CTSD) + (0.276 × IL3RA). The median risk score was used to categorize the patients in the training and validation cohorts into high- and low-risk categories. We discovered that the high-risk group in the TCGA training cohort had a lower survival rate (Fig. 5D).

Construction of an immune-related four-gene signature for cDC1s: A 93 immune-related genes in cDC1s were subjected to univariate Cox analysis (P < 0.05); B examining four immune-related genes in cDC1s using least absolute shrinkage and selection operator (LASSO) coefficients; C parameter adjustment in the LASSO model by a process of tenfold cross-validation; D, E analysis of the overall survival of TNBC patients in the high- and low-risk training and testing cohorts; F, G to confirm prognostic accuracy among TNBC patients, receiver operating characteristic (ROC) curves were used in both the training and testing populations
Similarly, in the GSE58812 validation cohort, the results for high-risk patients were significantly worse than those for low-risk individuals (Fig. 5E). We conducted ROC curve analysis in both the training and validation populations to further evaluate the risk score's predictive power for the prognosis of TNBC patients. The TCGA cohort's 1-, 3-, and 5-year survival AUC values were 0.804, 0.791, and 0.786, respectively, as shown in (Fig. 5F). Because there were only a few patients who were followed up for a year, we generated prognostic ROC curves for the validation cohort's 2-, 3-, and 5-year outcomes, yielding AUC values of 0.728, 0.692, and 0.756, respectively (Fig. 5G). These results indicated that the risk score has high accuracy in predicting patient outcomes among both cohorts.
3.5 Validation of prognostic cDC1 immune-related genes in training and testing datasets
Patients in the training group were categorized as high- or low-risk according to their median risk score for the purposes of the survival analysis. The death rate among TNBC patients was found to be proportional to their risk score (Fig. 6A), which was confirmed in the test group (Fig. 6B). Of note, CTSD and IL3RA showed noticeably greater expression levels in the training and testing datasets in the group at higher risk as opposed to the group with lower risk (Figs. 6C, D). These results imply that these cDC1 immune-related genes may serve as important predictors of adverse outcomes. Contrarily, the degrees of manifestation of IDO1 and HLA-DOB were substantially lower in the group at high risk than in the group at low risk, indicating that these specific cDC1 immune-related genes may possess a protective potential, as shown in Figs. 6C and D. The Kaplan–Meier analysis's findings further confirmed the close association of these four cDC1 immune-related genes with prognosis in patients with TNBC (Fig. 6E). Additionally, we contrasted the rates of survival of TNBC patients according to age, stage, and classifications of tumor (T), node (N), and metastasis (M). High-risk individuals continually had a worse overall survival rate compared to those at low risk (Fig. 7), suggesting that the model we established can be effectively utilized for a variety of clinical factors.

Verification of the cDC1 immune-related genes’ predictive significance in training and testing datasets: A, B point graphs showing the training and testing cohorts’ risk and survival ratings; C, D gene expression heatmap that compares the expression levels of four immune-related cDC1 genes between high- and low-risk groups in the training and testing cohorts; E Kaplan–Meier analysis is used to assess the impact of both high and low expression of these four cDC1 immune-related genes on the prognosis of TNBC patients

Kaplan–Meier survival curves for individuals with various clinical characteristics, divided into groups at low and high risk
3.6 The four-gene cDC1 immune-related signature's independent prognostic potential in TNBC patients
Both univariate and multivariate Cox regression experiments showed that the risk assessment score-based model was an independent predictor of TNBC patients (Fig. 8A). Of note, in contrast to other clinical characteristics, the risk score had an AUC value of 0.786, indicating remarkable predictive power (Fig. 8B).

Gene set enrichment analysis (GSEA) comparing high- and low-risk groups, as well as the four-gene cDC1 immune-related signature’s independent and better predictive abilities in TNBC patients: A using both univariate and multivariate techniques to analyze clinical characteristics and risk scores; B using risk ratings and additional clinical data to get area under the curve values; C GSEA has the most enriched pathways between the low- and high-risk categories
We used a GSEA to more thoroughly investigate the molecular variations between the high- and low-risk groups. The study's findings showed that the high-risk group's gene sets were more common in pathways related to drug metabolism including drug metabolism (cytochrome P450), drug metabolism (other enzymes), metabolism of xenobiotics by cytochrome P450, oxidative phosphorylation, and tyrosine metabolism, indicating that compared to patients in the low-risk group, individuals in the high-risk group showed more active metabolic processes especially those related to medication metabolism and energy production. In contrast, the low-risk group had more gene sets in immune system-related pathways including allograft rejection, autoimmune thyroid disease, graft-vs.-host disease, intestinal immune network for immunoglobulin A production, and systemic lupus erythematosus. Enrichment of these pathways suggested that compared to patients in the high-risk group, those in the low-risk group might have better immune responses and self-regulation skills (Fig. 8C).
3.7 Development of the prognostic nomogram
We created a nomogram that included age, clinical stage, T, N, and M categorization, and risk score in order to more accurately evaluate the risk in patients with TNBC (Fig. 9A). We generated calibration curves to further confirm this innovative nomogram's predictive power. The results showed that the nomogram's predictions were accurate, with the predicted 1-, 3-, and 5-year survival rates compared to the actual survival rates having a C-index of 0.927 (95% CI: 0.873–0.980) (Fig. 9B). We also conducted a decision curve study to assess the usefulness of clinical judgment by calculating the net benefit at each threshold probability for each clinical feature. The results of this analysis showed that the risk score outperformed other clinical indicators, suggesting that it is a good prognostic indicator for patients and can guide clinical decisions (Fig. 9C).

Development of the prognostic nomogram: A the nomogram was developed by combining clinical features and risk score; the TCGA-TNBC samples had 1-, 3-, and 5-year survival rates of 0.997, 0.929, and 0.791, respectively. *** P < 0.001; B model calibration curves with a C-index of 0.927 (95% CI: 0.873–0.980) for 1-, 3-, and 5-year survival periods. The X-axis represents the predicted probability of survival at specific time points as determined by the nomogram, while the Y-axis represents the observed probability of survival at corresponding time points in the validation dataset. C decision curve analysis to assess the prognostic model's clinical usefulness. The X-axis represents the threshold probability, which is the probability at which a patient would opt for treatment based on the model's prediction, while the Y-axis represents the net benefit, calculated as the difference between true-positive predictions and false-positive predictions, adjusted for the threshold probability
3.8 Tumor mutation characteristics and chemotherapy response in TNBC based on risk scores
Twenty genes in total showed the highest mutation frequencies in both risk categories, according to the results shown in the waterfall plot (Fig. 10A). Notably, the low-risk group had substantially greater mutation frequencies for these genes than the high-risk group. Furthermore, the results showed that TMB levels were higher in low-risk individuals than in high-risk ones (Fig. 10B). and that those with a lower TMB had a relatively shorter overall survival compared to those with a higher TMB (Figs. 10C, D), which is consistent with previous studies [35, 36]. The IC50 values were calculated to predict the chemotherapy response, the results of which indicated that the predictive response to docetaxel was better for the high-risk group, while the predictive response to paclitaxel was better for the group at low risk. However, the IC50 values for cisplatin, doxorubicin, and gemcitabine did not significantly differ between the two groups (Fig. 10E).

Based on risk score features, tumor mutations and chemotherapy response factors in TNBC: A the waterfall plot shows somatic genomic alterations in individuals with TNBC, divided into risk scores that are high (left) or low (right); B tumor mutation burden values for high- and low-risk patients; C survival analysis for the specified four groups; D survival rates in groups with a higher and lower load of tumor mutations; E contrasting the expected half-maximal inhibitory concentrations of chemotherapeutic drugs in patients at high and low risk
3.9 The potential of cDC1 immune-related features in anti-PD-1/L1 immunotherapy and variations in immune infiltration patterns among risk groups
The TNBC samples' immune cell composition was ascertained by characterizing their immune infiltration patterns using the CIBERSORT method. Notably, gamma delta (γδ) T cells, memory-activated CD4 + T cells, CD8 + T cells, and M1 macrophages were all more prevalent in the low-risk group, while M0 and M2 macrophages were much more prevalent in the high-risk group (Fig. 11A). After that, we calculated function scores for particular immune cells. The majority of immune cells and their roles varied significantly between patients at high and low risk, according to the results (Fig. 11B). To better understand these inequalities, we employed a heat map to look at any differences in clinical variables between the high- and low-risk groups. We found that age, staging, and N staging were significantly different (Fig. 11C). Six immune subtypes have been found in recent research based on the molecular subtype and type of cancer tissue including C1 (associated with wound healing), C2 (dominated by interferon gamma [IFN-γ]), C3 (characterized by inflammation), C4 (depleted of lymphocytes), C5 (immunologically silent), and C6 (dominated by transforming growth factor beta [TGF-β]) [37]. According to the study's findings, the high-risk TNBC subgroup had a greater prevalence of subtypes C1 and C3, which may be related to immunological responses and inflammation, whereas subtype C2 was more common indicating a more robust immune regulation function in the low-risk population (Fig. 11D). Additionally, the low-risk group displayed higher estimated and immunological ratings than the high-risk group (Fig. 11E). We utilized the IMvigor 210 database to analyze the responses of both risk groups to anti-PD-L1 treatment. The IMvigor 210 cohort's low-risk group had greater overall survival rates, per the Kaplan–Meier curve analysis (Fig. 11F). Risk score evaluations for complete/partial response and stable/progressive disease, however, did not show any appreciable variations (Fig. 11G). Additionally, we assessed the ICI response risk model's predictive potential and discovered that low-risk individuals had higher expression levels of the immune checkpoint markers CD274, CTLA4, LAG3, and PDCD1, suggesting that ICI treatment would work better for them (Fig. 11H).

Patterns of immune infiltration and the possibility of cDC1 immune-related characteristics in anti-PD-1/L1 immunotherapy across various risk groups: A CIBERSORT algorithm scores for 22 immune cell types (*P < 0.05, **P < 0.01, ***P < 0.001); B scores based on ssGSEA of 29 gene sets associated to immunotherapy (*P < 0.05, **P < 0.01, ***P < 0.001); C clinical characteristics of high- and low-risk groups (*P < 0.05, **P < 0.01); D correlation analysis between the risk model and immunological phenotypes (chi-squared test: P = 0.002); E tumor microenvironment scores for high- or low-risk patients (**P < 0.01, ***P < 0.001); F overall survival (OS) of IMvigor210 database individuals treated with anti-PD-L1 who were at high or low risk; G risk ratings for patients who obtained SD/PD or CR/PR (full response, partial response, stable disease, progressive disease, and SD/PR, respectively); H immune checkpoint expression and its association with Spearman correlation-based risk ratings
4 Discussion
TNBC often leads to poor clinical outcomes owing to the absence of effective targeted therapies and the propensity for resistance to treatment [3]. However, the advent of immunotherapy has provided a powerful clinical approach to cancer therapy, with several studies highlighting its potential in improving survival rates in TNBC patients [38, 39].The immunogenic characteristics of TNBC make it more likely to benefit from immunotherapy. However, most patients exhibit low effective response rates to these treatments [40]. Therefore, it is crucial to identify potential immunotherapeutic targets that depict the complex relationships that exist between tumor cells and the immune system. Previous studies involving several preclinical cancer models have highlighted the significant role of cDC1s in effective immunotherapy. Furthermore, the degree of cDC1 infiltration is linked to a better prognosis and response to immunotherapy in human malignancies [41]. Previous studies have indicated that in non-small cell lung cancer (NSCLC), CCL7 promotes T cell expansion by recruiting cDC1 to the TME, thereby enhancing the efficacy of anti-PD-1 therapy in NSCLC mouse models [42]. Similarly, in the treatment of advanced pancreatic cancer, Rintatolimod administration was associated with an increased peripheral abundance of cDC1 cells, which correlated with improved clinical outcomes [43]. However, the application of cDC1 in TNBC immunotherapy remains unexplored. Given the pivotal role of cDC1 in shaping the tumor immune microenvironment, further investigation is warranted to elucidate its therapeutic potential and develop effective strategies for TNBC treatment. Therefore, we acquired and processed the GEO database's scRNA-seq data with which to conduct a comprehensive analysis of both patients with TNBC and normal breast tissue, and constructed a detailed map of cell composition in the TME of TNBC. We identified specific DEGs in cDC1s through differential gene analysis, after which to find predictive genes, we used LASSO and univariate Cox regression analysis on data from the TCGA-TNBC cohort. A prognostic model was developed using four immune-related genes (IDO1, HLA-DOB, CTSD, and IL3RA) associated with cDC1s and the accuracy of this four-gene signature was subsequently validated. Of note, these four genes were found to be closely related not only to prognosis, but also to the TME of TNBC, tumor mutation characteristics, chemotherapy response, and immune treatment response. Our research advances our knowledge of TNBC tumor immunology and offers fresh ideas for future treatment approaches meant to enhance patient outcomes.
The essential enzyme indoleamine 2,3-dioxygenase 1 (IDO1), which catalyzes the breakdown of tryptophan and the accumulation of kynurenine, is how DCs control tolerance [44]. When expressed in DCs, IDO1 exerts potent immunoregulatory effects [45]. IDO1-mediated tryptophan metabolism is among the metabolic pathways that have been investigated the most in relation to immunological tolerance and tumor cell immune evasion, representing a key mechanism underlying tumor resistance to CTLA-4 or anti-PD-1 therapies [46]. This suggests, therefore, that IDO1 targeting could be used as a possible supplement to the TNBC immunotherapy now in use. Additionally, HLA-DOB is one of the two components (β chain) of HLA-DO, a human leukocyte antigen class II heterodimer that regulates peptide loading onto MHC-II molecules mediated by HLA-DM and acts as an antigen presentation modulator. Previous studies have indicated that MHC-II interactions can induce the CD40 signaling necessary for ideal CD8 + T cell responses, strengthening protection against tumors [17]. Studies have shown that the cathepsin D (CTSD) gene is closely related to the clinical malignancy and prognosis of glioma [47]. In colorectal cancer liver metastasis, CTSD is considered to be a potential key N-glycosylated protein and affects the invasion and metastasis of CRC cells by cleaving ACADM under the regulation of glycosyltransferase complexes DDOST and STT3B [48]. IL3RA (CD123), a cell membrane protein called the alpha subunit of the interleukin-3 receptor is overexpressed in a number of hematological malignancies including acute myeloid leukemia and plasmacytoid DC neoplasms [49]. Additionally, it controls hematopoietic cell differentiation, survival, and proliferation. However, CTSD and IL3RA are not extensively explored as therapeutic targets in the context of TNBC immunotherapy. Our findings highlight their potential relevance, offering a novel perspective for advancing immunotherapeutic strategies in TNBC. Nevertheless, further in-depth experimental investigations and clinical validation are imperative to substantiate these findings and fully elucidate their therapeutic implications.
Immune cell infiltration analysis is crucial for both the diagnosis and treatment of diseases. Therefore, in this investigation, the immunological status was thoroughly examined among different risk groups of patients with TNBC. Tumor phenotypes can be categorized into two main patterns based on immune cell infiltration: “hot” and “cold,” correlating with favorable and unfavorable responses to anti-tumor immunotherapy, respectively [50]. Of note, the high-risk group exhibited increased presence of M0 and M2 macrophages with lower immune scores, classifying them as “cold” tumors. Conversely, the low-risk group displayed higher abundances of CD8 + T cells, memory-activated CD4 + T cells, γδ T cells, and M1 macrophages, indicating a “hot” tumor phenotype. Furthermore, immune checkpoint molecule expression is higher in low-risk patients, suggesting a better response to ICI. Existing evidence links increased PD-L1 expression with enhanced immunotherapy effectiveness [51], a conclusion that was further supported by IMvigor 210 database analysis. The sensitivity of the high- and low-risk groups to popular chemotherapeutic drugs, such as docetaxel and paclitaxel, varied. The safety and effectiveness of combining chemotherapy with ICI have been established by earlier research [8, 9]. The findings of this study indicated that low-risk patients had higher TMB, a biomarker for assessing the efficacy of immunotherapy [52], compared to high-risk individuals, indicating a higher chance that immunotherapy will benefit low-risk. These results are consistent with those of other clinical studies showing more robust responses in patients with a high TMB [35, 36]. Additionally, we looked at the correlation between TMB and risk ratings and discovered that low-risk patients had more TP53, PIK3CA, and HMCN1 genes than high-risk patients. These mutation characteristics can help choose immunotherapies for certain patients more precisely.
This study used four cDC1 immune-related genes to create a novel prognostic model, revealing for the first time the potential functions of cDC1-specific genes in the TME through single-cell sequencing. We have also introduced novel molecular markers that provide fresh perspectives for the clinical management and immunotherapy treatment of patients with TNBC. This model not only elucidated the TNBC immunological microenvironment's intricacy, but also offered significant references for patient prognostication and laid the theoretical groundwork for personalized medicine.
5 Limitation
This study had a few limitations. First, the lack of experimental validation meant that we could not confirm the specific roles of the four selected genes in TNBC through functional experiments, which introduces uncertainty regarding their biological mechanisms and clinical relevance. Second, the reliance on publicly available datasets may have introduced selection bias, as the samples included might not fully represent the heterogeneity of TNBC across different populations. Third, potential confounding factors, such as variations in the tumor microenvironment, patient treatment histories, and sequencing techniques, may have influenced the analysis and model performance, necessitating caution in interpreting the results. Finally, insufficient clinicopathological data limited the comprehensive assessment of the predictive capacity of risk features; therefore, additional experimental and clinical studies are needed to further confirm our findings, confirm their potential in practical applications, and deepen our understanding of the roles of these genes in TNBC.
6 Conclusion
In summary, this study established and externally validated a novel prognostic model based on four cDC1 immune-related genes (IDO1, HLA-DOB, CTSD, and IL3RA) through comprehensive analysis of bulk RNA and scRNA-seq data from TNBC patients. This model provides insights into the immunological characteristics of TNBC and offers potential tools for risk stratification and prognostication. These findings may contribute to advancing personalized treatment strategies for TNBC and guiding clinical immunotherapy decisions.
However, the study has several limitations that should be addressed. First, the roles of these four genes in the tumor immune microenvironment have not been experimentally validated, making it challenging to establish their precise biological mechanisms and clinical utility. Second, the limited availability of clinicopathological data restricted a more robust assessment of the model's predictive performance. Future research should focus on functional validation of these genes through in vitro and in vivo experiments and involve larger, multicenter cohorts to evaluate the model's applicability in clinical practice. These efforts will be crucial for translating our findings into actionable insights for TNBC treatment.
Data availability
All data used in this study are derived from publicly available summary data from the GEO databases and TCGA databases. Data generated during this study are available from the corresponding author upon reasonable request.
References
Brown M, Tsodikov A, Bauer KR, et al. The role of human epidermal growth factor receptor 2 in the survival of women with estrogen and progesterone receptor-negative, invasive breast cancer: The California cancer registry, 1999–2004. Cancer. 2008;112(4):737–47.
Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019;9(2):176–98.
Denkert C, Liedtke C, Tutt A, et al. Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. The Lancet. 2017;389(10087):2430–42.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: towards combination strategies with curative potential. Cell. 2015;161(2):205–14.
Luen S, Virassamy B, Savas P, et al. The genomic landscape of breast cancer and its interaction with host immunity. Breast. 2016;29:241–50.
Loi S, Drubay D, Adams S, et al. Tumor-infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early-stage triple-negative breast cancers. J Clin Oncol. 2019;37(7):559–69.
Gatalica Z, Snyder C, Maney T, et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomark Prev. 2014;23(12):2965–70.
Schmid P, Cortes J, Pusztai L, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382(9):810–21.
Schmid P, Rugo HS, Adams S, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(1):44–59.
Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37(4):443–55.
Böttcher JP, Reis E, Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. 2018;4(11):784–92.
Broz ML, Binnewies M, Boldajipour B, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen presenting cells, critical for t cell immunity. Cancer Cell. 2014;26(5):638–52.
Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8a+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–100.
Sánchez-Paulete AR, Cueto FJ, Martínez-López M, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires Batf3-dependent dendritic cells. Cancer Discov. 2016;6(1):71–9.
Barry KC, Hsu J, Broz ML, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. 2018;24(8):1178–91.
Michea P, Noël F, Zakine E, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. 2018;19(8):885–97.
Ferris ST, Durai V, Wu R, et al. cDC1 prime and are licensed by CD4 T cells to induce anti-tumour immunity. Nature. 2020;584(7822):624–9.
Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of t cell immunity in melanoma. Cancer Cell. 2016;30(2):324–36.
Garris CS, Arlauckas SP, Kohler RH, et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49(6):1148-1161.e7.
Hubert M, Gobbini E, Couillault C, et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci Immunol. 2020;5(46):eaav3942.
Ruffell B, Chang-Strachan D, Chan V, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–37.
Wu SZ, Al-Eryani G, Roden DL, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53(9):1334–47.
Pal B, Chen Y, Vaillant F, et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021;40(11): e107333.
Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets—update. Nucl Acids Res. 2012;41(D1):D991–5.
Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):A68–77.
Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):15.
Zhang X, Lan Y, Xu J, et al. Cell Marker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47(D1):D721–8.
Domínguez Conde C, Xu C, Jarvis LB, et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science. 2022;376(6594):eab15197.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes agenomes. Nucleic Acids Res. 1999;27(1):29–34.
Wu T, Hu E, Xu S, et al. ClusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. 2021;2(3):100141.
Iasonos A, Schrag D, Raj GV, et al. How to build and interpret a nomogram for cancer prognosis. J Clin Oncol. 2008;26(8):1364–70.
Mayakonda A, Lin DC, Assenov Y, et al. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747–56.
Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE. 2014;9(9): e107468.
Dusenbery AC, Maniaci JL, Hillerson ND, et al. MHC class I loss in triple-negative breast cancer: a potential barrier to PD-1/PD-L1 checkpoint inhibitors. Am J Surg Pathol. 2021;45(5):701–7.
Negrao MV, Skoulidis F, Montesion M, et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J Immunother Cancer. 2021;9(8): e002891.
Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–6.
Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812-830.e14.
Yang F, Xiao Y, Ding JH, et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35(1):84-100.e8.
Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. J Natl Compr Canc Netw. 2020;18(4):479–89.
Mittendorf EA, Zhang H, Barrios CH, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396(10257):1090–100.
Cancel JC, Crozat K, Dalod M, et al. Are conventional type 1 dendritic cells critical for protective antitumor immunity and how? Front Immunol. 2019;10:9.
Zhang M, Yang W, Wang P, et al. CCL7 recruits cDC1 to promote antitumor immunity and facilitate checkpoint immunotherapy to non-small cell lung cancer. Nat Commun. 2020;11(1):6119.
Van Eijck CWF, Haddaoui HE, Kucukcelebi S, et al. Rintatolimod in advanced pancreatic cancer enhances antitumor immunity through dendritic cell-mediated T-cell responses. Clin Cancer Res. 2024;30(16):3447–58.
Munn DH, Shafizadeh E, Attwood JT, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–72.
Sharma MD, Pacholczyk R, Shi H, et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity. 2021;54(10):2354-2371.e8.
Holmgaard RB, Zamarin D, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210(7):1389–402.
Zheng W, Chen Q, Wang C, et al. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol Carcinog. 2020;59(6):651–60.
Xiong N, Du Y, Huang C, et al. N- glycosylation modification of CTSD affects liver metastases in colorectal cancer. Adv Sci. 2024. https://doi.org/10.1002/advs.202411740.
Muñoz L, Nomdedéu JF, López O, et al. Interleukin-3 receptor α chain (CD123) is widely expresssed in hematologic malignancies. Haematologica. 2001;86(12):1261–9.
Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Disc. 2019;18(3):197–218.
Zhang S, Zhang W, Zhang J. 8-Gene signature related to CD8+ T cell infiltration by integrating single-cell and bulk RNA-sequencing in head and neck squamous cell carcinoma. Front Genet. 2022;13: 938611.
Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348(6230):124–8.
Acknowledgements
The authors express their gratitude to the GEO database, and the TCGA database for their contributions and the researchers involved in sharing the data.
Funding
This study was funded by National Natural Science Foundation of China (82430123, 82405170), Shandong Province Traditional Chinese Medicine Technology Project (Q-2023098) and The “Taishan Scholars” Special Foundation of Shandong Province (tstp20221166).The funding bodies exerted no influence over the study design, data collection, analysis, interpretation, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
Study design: C.G.S. and H.Y.L.; Data extraction and analyses: L.N.H.; Results presentation and interpretation: Y.R.L.; Manuscript review and editing: Y.L.S.andQ.S.; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Institutional review board statement
Not applicable. All data used in this study were derived from publicly available datasets, which had received approval from the relevant ethics committees. No human data were collected in this study; therefore, further ethical approval was not necessary.
Informed consent
Not applicable. No human data were collected in this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, L., Liu, Y., Shi, Y. et al. Comprehensive single-cell analysis of triple-negative breast cancer based on cDC1 immune-related genes: prognostic model construction and immunotherapy potential. Discov Onc 16, 206 (2025). https://doi.org/10.1007/s12672-025-01929-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-025-01929-1