
Abstract
Pure erythroid leukemia (PEL) is a rare, aggressive subtype of acute myeloid leukemia with a poor prognosis. The diagnosis of PEL is often medically urgent, quite challenging, and is typically a diagnosis of exclusion requiring meticulous distinction from non-neoplastic erythroid proliferations, particularly florid erythroid hyperplasia/regeneration. Given the frequency of TP53 mutations in the molecular signature of PEL, we hypothesize that differential p53 expression by immunohistochemistry (IHC) may be useful in distinguishing PEL versus non-neoplastic erythroid conditions. We performed p53 IHC on 5 normal bone marrow, 46 reactive erythroid proliferations, and 27 PEL cases. We assessed the positivity and intensity of nuclear staining in pronormoblasts and basophilic normoblasts using a 0–3+ scale with 0 being absent (with internal positive controls) and 3 being strong nuclear positivity. A total of 26/27 PEL cases showed strong, uniform, diffuse intense staining by the neoplastic pronormoblasts versus 0/5 and 0/46 normal and reactive controls, respectively. The control cases show various staining patterns ranging from 0 to 3+ in scattered erythroid precursor cells. Uniform, strong p53 positivity is unique to PEL and discriminates this entity from a benign erythroid mimic. Thus, p53 IHC may be a useful marker in urgent medical cases to assist in the confirmation of a malignant PEL diagnosis while awaiting the results of additional ancillary studies such as cytogenetics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pure erythroid leukemia (PEL) (previously known as acute myeloid leukemia (AML)-M6b) is a rare, aggressive subtype of AML with poor prognosis. According to the current revised fourth edition WHO criteria, PEL is defined as a neoplasm with > 80% erythroid lineage cells in the bone marrow (BM) and ≥ 30% proerythroblasts, with no significant myeloblast component [1, 2].
The diagnosis of PEL is often quite challenging, as it is typically a diagnosis of exclusion and requires meticulous distinction from non-neoplastic erythroid proliferations, particularly florid erythroid hyperplasia/regeneration. To establish a diagnosis of PEL, erythroid lineage cell counts are determined based on morphology in a BM aspirate smear. From a purely morphologic perspective, the cellular makeup of PEL is variable across cases and overlaps what is seen in non-neoplastic erythroid proliferations of various causes, including megaloblastic anemia, hemoglobinopathies, autoimmune hemolytic anemia, and florid erythroid regeneration after BM injury/insult [3].
While certain erythroid lineage-associated immunohistochemical (IHC) markers exist (e.g., hemoglobin A, glycophorin, alpha hemoglobin stabilizing protein, and GATA1) [4,5,6,7], these may not be routinely available in all pathology practices. Therefore, establishment of erythroid lineage may rely on a combination of other markers such as CD117, CD71, and E-cadherin [8, 9], along with demonstrating the lack of other lineage-defining antigens (e.g., myeloperoxidase, cytokeratins, plasma cell, B-cell, and T-cell markers). Additionally complicating the diagnosis, hemoglobin A and glycophorin are commonly negative in the predominantly pronormoblastic proliferations of PEL [9]. Along with the complexity of establishing erythroid lineage, there is not usually a simple immunophenotypic way to discriminate benign from malignant proliferations within the erythroid lineage. As such, when dealing specifically with the issue of distinguishing a benign erythroid proliferation versus PEL, identification of alternate IHC markers which may contribute to making this distinction could be useful for pathologists.
One characteristic feature of PEL is maturation arrest of the erythroid lineage leading to elevated numbers of pronormoblasts and other left-shifted erythroid precursors in the BM [8, 10]. While the specific causes of maturation arrest and failure of pronormoblast differentiation in PEL is largely unknown, p53 has been demonstrated to play an important role in promoting cell cycle arrest in hematopoietic stem cells [11]. Additionally, TP53 gene deletions and/or mutations are considered a characteristic molecular feature in PEL and are seen in over 90% of cases [12].
Given the frequency of TP53 mutations in the molecular signature of PEL, we hypothesize that strong p53 expression by IHC may be useful in supporting the presence of neoplastic erythroid precursors (e.g., PEL) versus weak or absent p53 expression in non-neoplastic erythroid conditions. As such, our study seeks to establish the utility of p53 expression by IHC in discriminating PEL from florid reactive erythroid proliferations of various causes.
Materials and methods
Case identification
We retrospectively searched our bone marrow pathology database from 2003 to 2020 for cases of pure erythroid leukemia (PEL). To be included in the study, each of these cases had to have available bone marrow slides for review and had to have a re-confirmed diagnosis of PEL according to current WHO classification diagnostic criteria [1] by an experienced hematopathologist (KKR). For the reactive erythroid proliferation comparison group, we searched our bone marrow pathology database from 2012 to 2018 for cases with the terms “erythroid hyperplasia” or “erythroid predominance” reported in the final diagnosis. We identified cases with a clinically annotated and morphologically documented marked reactive erythroid hyperplasia. Cases with concurrent or prior involvement of the bone marrow by a myeloid neoplasm, lymphoma, plasma cell proliferative disorder, and from patients that were post-hematopoietic stem cell transplantation were excluded. A thorough review of the clinical history was performed to document, when possible, the specific etiology for the erythroid hyperplasia. Finally, normal bone marrows (performed for the purpose of lymphoma staging) were selected from the bone marrow pathology archives.
Immunohistochemistry
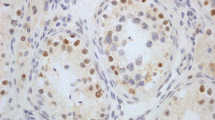
Immunohistochemical analysis was performed on 3–4-μm-thick, formalin-fixed, paraffin-embedded, decalcified bone marrow biopsy sections in all cases. A primary antibody to P53 (Ventana, clone DO-7) was utilized. Each case was scored independently in a blinded fashion by two authors (CAA and KKR) with an interrater reliability of 94.7%; discrepancies in scores were reviewed together and consensus was reached. Cases were scored for predominant intensity of staining in pronormoblasts (large nuclear size with 2–3 elongated nucleoli and dispersed chromatin) and basophilic normoblasts (0: negative, 1+: weak, 2+: moderate, 3+: strong) (as shown in Fig. 1), because it is very uncommon to see any degree of significant staining in the more terminally differentiated red blood cell forms (small nuclear size and deeply condensed chromatin obscuring any possible nucleoli) in benign cases of erythroid hyperplasia. In addition, most cases of PEL show minimal maturation beyond the pronormoblast/very early erythroid precursor stage; therefore, we aim to assess the percent positivity and degree of intensity in these specific cell types. In each case, the presence of at least five positive nuclei was required to ascertain that the stain was technically optimal.

p53 immunohistochemistry grading 0–3+ in bone marrow core biopsy pronormoblasts. The nuclear staining intensity in this photomicrograph illustrates the range of p53 immunohistochemical staining intensity that was evaluated and utilized in this study in early erythroid precursors. The arrows highlight an example of each cell staining pattern from 0 (no staining) to 3+ (strong staining) (p53 immunohistochemistry, bone marrow core biopsy, 1000× magnification)
Cytogenetics
Giemsa-banded (G-banded) chromosome analysis was performed on bone marrow samples according to conventional methods. When available, at least 20 metaphases were analyzed. Karyotypes of G-banded chromosomes were described according to the 2020 International System of Human Cytogenetic Nomenclature [13]. Abnormal clones were defined as two or more cells with the same structural abnormality, the same extra chromosomes, or the presence of three or more cells with loss of the same chromosome. Although technically clonal, cases with −Y, −X, + 15, and +Y were not considered a pathologic clonal finding but regarded as an age-related phenomenon. A complex karyotype is defined as > 3 structural and/or numerical abnormalities.
Next-generation sequencing
Targeted NGS was performed to detect gene mutations commonly found in myeloid hematologic malignancies as described previously [14, 15]. NGS testing was performed at several different institutions, and while the NGS panels were variable at each institution, all panels interrogated a minimum of 35 commonly mutated genes, including TP53.
Results
Clinical and morphologic findings
Pure erythroid leukemia
Twenty-seven cases of PEL were included. The cases included 19 males and 8 females, with a median age of 66 (age ranged from 26 to 78 years). All of these cases showed a striking erythroblastic proliferation consisting of a prominent (greater than 30%) population of pronormoblasts and met the current WHO diagnostic criteria [1]. Figure 2 demonstrates the typical bone marrow aspirate and bone marrow core biopsy morphologic features from two cases included in the study.

Cytologic features of exuberant reactive erythroid proliferations and pure erythroid leukemia (PEL) in bone marrow. The black lines in this composite photomicrograph demarcate six individual cases with the aspirate depicted on the left side and the core biopsy on the right side of each case. The upper and middle rows represent reactive disorders—upper row both cases vitamin B12 deficiency; the middle row left side autoimmune hemolytic anemia and the middle row right side beta thalassemia major hemoglobinopathy. The bottom row represents two cases of PEL. In the aspirate smears, a left-shifted, variable predominance of early erythroid precursors/pronormoblasts is evident. These cells are large, with a high nuclear/cytoplasmic ratio, round nuclear contours, 2–3 nucleoli, and deep blue cytoplasm. In the case of PEL, these neoplastic pronormoblasts may harbor cytoplasmic vacuoles. In general, the reactive cases tend to show some degree of continued background progressive erythroid maturation compared with PEL but in some instances, such as B12 deficiency, this may not be the case. In the corresponding bone marrow core biopsies, one can see similar degrees of left shift in the reactive cases with variable numbers of pronormoblasts. In the biopsy sections, pronormoblasts are recognized by their large size, cleared-out, stippled chromatin, 1–3 elongated nucleoli, and crisply-defined nuclear membranes. As in the aspirate smears, one may see a more uniform proliferation of early erythroid precursors in the core biopsy in a benign disorder, mimicking PEL (depicted here in B12 deficiency—upper row) (bone marrow aspirate, Wright-Giemsa stain, 1000× magnification; bone marrow core biopsy, Hematoxylin & Eosin, 1000× magnification)
Reactive erythroid proliferations
Forty-six reactive erythroid cases were identified. The etiologies of the erythroid hyperplasia were diverse and ranged across a variety of underlying reactive conditions (see Table 1 for a complete listing). The majority of our cases stemmed from a hemolytic condition, hemoglobinopathy, or megaloblastic anemia due to B12 deficiency. The cases included 25 males and 21 females, with median age of 55. These cases by morphology showed a predominant erythroid proliferation with a full spectrum of maturation and variable degree of left shift. No significant dysplasia in any of the lineages, increase in blasts, or population of abnormal monocytic cells was present. Figure 2 demonstrates the range of bone marrow aspirate and bone marrow core biopsy morphologic features from four cases included in this study. These four cases included two cases of B12 deficiency, autoimmune hemolytic anemia, and beta thalassemia major hemoglobinopathy.
Normal bone marrow
Five bone marrow cases, performed for the purpose of staging lymphoma, constituted our normal control cases. These five cases were from two males and three females, with a median age of 65.
p53 immunohistochemistry
Normal bone marrow
Normal bone marrow controls demonstrated predominantly 0–1+ intensity staining in pronormoblasts. Rare 2+ intensity staining was seen in two of the five normal controls, with no instances of 3+ intensity of staining in any of the five normal controls. Due to the normal, non-reactive state of these bone marrows, there were relatively few clusters of early erythroid precursor cells, as would be expected. Figure 3a demonstrates an example from one of the control bone marrows with the range of 0–2+ p53 positivity.

Comparison of p53 immunohistochemistry staining in bone marrow core biopsies in non-neoplastic erythroid proliferations and pure erythroid leukemia (PEL). This composite photomicrograph (viewed sequentially from a to i) depicts an increasing, progressive degree of background p53 nuclear staining that may be seen in normal staging bone marrows (a) and a range of reactive erythroid predominant disorders (b–h). The p53 staining is seen in scattered cells and, despite the heterogeneity on a case-by-case basis, is not uniform and diffusely strong but rather shows a subset of cells ranging from 0 to 3+/3+. The reactive conditions illustrated include hereditary sideroblastic anemia (b; 0+), sickle cell disease (c; mostly 1+), beta thalassemia minor (d; mostly 1+, rare 2+), autoimmune hemolytic anemia (e; mostly 1+, occasional 2+), vitamin B12 deficiency (f; mostly 1+, occasional 2+, occasional 3+), and vitamin B12 deficiency and beta thalassemia major (g, h, respectively; admixture of mostly 1+ and 2+ with occasional 3+). This variable staining pattern is distinctly different from the strong, diffuse, and uniform positivity that is seen in PEL (i) (graded as 3+/3+) (p53 immunohistochemistry; bone marrow core biopsy; 600× magnification)
Reactive erythroid proliferations
In the 46 cases of reactive erythroid proliferations, a spectrum of p53 staining patterns was identified and is detailed in Table 1. Importantly, we saw a range from essentially absent staining (aside from the required scattered internal positive control cells) to more robust staining with occasional, scattered 2+, and 3+ positive cells. This spectrum of positivity showing a gradation from minimal staining to increased staining is shown in Fig. 3 as one proceeds from Fig. 3b to h. Essentially absent staining was observed in the case of hereditary sideroblastic anemia (Fig. 3b) and case of liver failure (not shown). Predominantly negative staining with rare and/or weakly staining erythroid precursors was observed in five cases. Sickle cell disease, as an example, is shown in Fig. 3c. In 17 cases, the predominant pattern was 1+. Figure 3d is from a case of beta thalassemia minor illustrating this 1+ pattern. In 12 cases, there was an admixture of 1+ and 2+ staining. Typical examples of such patterns are shown in Fig. 3e and f from cases of autoimmune hemolytic anemia and vitamin B12 deficiency, respectively. For the remaining 10 cases, there was a range from 1 to 3+, with the most substantial 3+ staining seen in cases 32 and 23 representing vitamin B12 deficiency and beta thalassemia major (Fig. 3g and h), respectively. Importantly, of these 46 cases demonstrating reactive erythroid and often left-shifted proliferations, we did not identify any cases with uniform/diffuse 2+ or 3+ intensity.
Pure erythroid leukemia
Compared with the reactive erythroid proliferation controls, 26/27 (96%) PEL cases had > 95% staining of erythroid precursors with strong 3+ intensity. This staining pattern is clearly distinct and unique compared with all of the normal bone marrow and reactive erythroid cases that were evaluated in this study. This dramatic, uniform, and strong, diffuse intense staining is illustrated in Fig. 3i. Interestingly, we did identify one PEL case (4%) that was clearly negative for p53 immunohistochemical staining with scattered positive internal controls demonstrating that the stain was indeed technically optimal.
Cytogenetic and next-generation sequencing findings
Reactive erythroid proliferations
Of the reactive erythroid proliferation controls, 35 of 46 had a conventional cytogenetic study performed. Thirty-three of these cases were karyotypically normal, and 2 cases harbored constitutional abnormalities (summarized in Table 1). None of the cases had next-generation sequencing performed, as this testing was either not available and/or not indicated.
Pure erythroid leukemia
All 27 cases exhibited complex karyotypes with both numerical and structural abnormalities (data not shown), as would be expected based on published literature. Twenty-one of the 27 cases (78%) demonstrated a cytogenetic and/or molecular genetic (NGS) abnormality involving chromosome 17p13/TP53. The details of these particular genetic abnormalities are shown in Table 2. As the karyotypes in PEL are complex (often highly complex with more than 10 structural and numerical abnormalities), it may be challenging within such a karyotype to clearly discern the presence or absence of a TP53 deletion event. Not all cases showed a clear-cut chromosome 17 abnormality by karyotype, as was the case in 10 of the 27 cases. In six of these cases, neither fluorescence in situ hybridization for a TP53 deletion nor next-generation sequencing studies were performed. Importantly, 4 of those 10 cases with no karyotypic abnormality on chromosome 17 did demonstrate a TP53 mutation by next-generation sequencing. Overall, eight of the PEL cases had next-generation sequencing data available, and all eight of these cases demonstrated TP53 mutations by NGS. In the single case of PEL with negative p53 IHC (case 27), monosomy 17 was present by karyotype. Next-generation sequencing was not available at the time of this diagnosis.
Discussion
Herein, we report the novel finding that strong and diffuse p53 immunohistochemical staining aids as a discriminator of pure erythroid leukemia (PEL) from otherwise exuberant, non-neoplastic, erythroid proliferations. The findings from our study demonstrate that significantly strong and uniform p53 IHC positivity in early erythroid precursors (pronormoblasts and basophilic normoblasts) correlate with a diagnosis of PEL as compared with negative to moderate and variable staining in cases of non-neoplastic erythroid proliferations. Reactive erythroid hyperplasia is a well-known morphologic mimic of PEL in many diverse clinical situations due to erythroid hyperplasia and left-shifted maturation. Although cytogenetic and molecular studies may ultimately resolve the differential diagnosis, these cases are often diagnostically time-sensitive due to the underlying critical condition(s) of patients. Therefore, this additional, widely available ancillary tool, p53 IHC, can serve as a rapid diagnostic aid and may even help guide ancillary testing.
Of note, we did identify a single case of PEL (1/27) that was negative for p53 by immunohistochemistry. This is an important take-away point because the lack of strong and diffuse p53 IHC (although rare based on our experience in this study) does not necessarily unequivocally exclude the possibility of PEL. However, the converse staining pattern of strong, diffuse p53 staining appears to be sufficient to exclude non-neoplastic, reactive erythroid-dominant hyperplasia. Possible explanations for the negative staining in the neoplastic cells could include a mutation that results in decreased/absent production of p53 protein, a mutation that renders rapid intracellular degradation of the p53 protein or an alteration in the epitope of the p53 protein such that it is not recognized/bound by the p53 antibody.
It is interesting to note that of the PEL cases, 21/27 had at least one p53 abnormality, seen either as chromosome 17p13 deletion or TP53 mutation detected by NGS, or both. Ongoing studies of TP53 mutations and the role of mutant p53 forms in tumorigenesis are beginning to elucidate the complex nature of its activity and expression. In a study of myelodysplastic syndrome, Saft et al. report that bone marrow progenitor cells which demonstrated strong p53 expression as detected by immunohistochemistry (IHC) were found to have missense mutations in TP53 [16]. A review of p53 mutant activity and regulation reveals growing evidence for gain-of-function TP53 missense mutations involved in tumorigenesis; these mutations are associated with accumulation of p53 variants within tumor cells, which can be identified by IHC [17, 18]. In contrast, benign cells are likely to express the wild-type p53 protein, which is maintained at relatively low levels in the cell by regulatory systems, including Mdm2 and Mdm4 which play a role in p53 inactivation and degradation [19].
Based on the findings in our study and current scientific literature, we hypothesize that strong p53 expression in PEL, in comparison with low-to-moderate expression levels in reactive erythroid hyperplasia, indicates the presence of a pathogenic variant of p53, which is more likely to accumulate in cells than the wild-type form. In future studies, it may be useful to identify the TP53 mutations associated with PEL and explore the relationship between mutational status and p53 expression.
We acknowledge that several limitations exist from this study. First, it is important to note that our findings apply solely to the discrimination of malignant pure erythroid leukemia cases as compared with benign erythroid lineage proliferations. The p53 IHC staining pattern in all other acute and chronic myeloid malignancies that may have an increased number of erythroid precursors has not been addressed. Second, our study includes a relatively small number of total tested cases. While the total number of reactive erythroid proliferations in this study is limited at 46, we do think that the wide variety of underlying conditions and the finding of occasional cases with more exuberant staining are likely representative of what is to be encountered in routine clinical practice. Third, it should be recognized that implementation of a nuclear immunostain in decalcified bone marrow specimens can sometimes be technically challenging. In terms of p53 immunohistochemistry, specific attention should be made to the particular clone that is utilized and that appropriate internal positive controls are present (albeit may be minimal) to ensure the validity of the assay and that there is indeed appropriate immunoreactivity and subsequent pathologic interpretation.
PEL is a difficult diagnosis, typically one of exclusion from an otherwise exuberant reactive erythroid proliferation. In this study, we show that strong and uniform p53 immunohistochemical positivity in early erythroid precursors is of diagnostic utility in situations requiring a prompt presumptive diagnosis of pure erythroid leukemia. Future studies aimed at identifying TP53 mutations in PEL and exploring the relationship between mutational status and the expression of p53 may be of interest and could potentially identify a candidate for p53-directed targeted therapy.
Data availability
Not applicable.
References
Arber DA, Brunning RD, Orazi A (2017) Pure erythroid leukemia. In: Swerdlow S, Campo E, Harris N et al (eds) WHO Classification of Tumours of Haematopoeitic and Lymphoid Tissues. IARC, Lyon, pp 161–162
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–2405. https://doi.org/10.1182/blood-2016-03-643544
Wang SA, Hasserjian RP (2015) Acute erythroleukemias, acute megakaryoblastic leukemias, and reactive mimics. Am J Clin Pathol 144(1):44–60. https://doi.org/10.1309/ajcprkyat6ezqhc7
Raess PW, Paessler ME, Bagg A, Weiss MJ, Choi JK (2012) α-Hemoglobin-stabilizing protein is a sensitive and specific marker of erythroid precursors. Am J Surg Pathol 36(10):1538–1547. https://doi.org/10.1097/PAS.0b013e31825fa501
Yu H, Pinkus JL, Pinkus GS (2016) α-Hemoglobin-stabilizing protein: an effective marker for erythroid precursors in bone marrow biopsy specimens. Appl Immunohistochem Mol Morphol 24(1):51–56. https://doi.org/10.1097/pai.0000000000000139
Lee WY, Weinberg OK, Pinkus GS (2017) GATA1 is a sensitive and specific nuclear marker for erythroid and megakaryocytic lineages. Am J Clin Pathol 147(4):420–426. https://doi.org/10.1093/ajcp/aqx018
Wolgast LR, Cannizzarro LA, Ramesh KH, Xue X, Wang D, Bhattacharyya PK, Gong JZ, McMahon C, Albanese JM, Sunkara JL, Ratech H (2011) Spectrin isoforms: differential expression in normal hematopoiesis and alterations in neoplastic bone marrow disorders. Am J Clin Pathol 136(2):300–308. https://doi.org/10.1309/ajcpsa5rnm9igfjf
Reinig EF, Greipp PT, Chiu A, Howard MT, Reichard KK (2018) De novo pure erythroid leukemia: refining the clinicopathologic and cytogenetic characteristics of a rare entity. Mod Pathol 31(5):705–717. https://doi.org/10.1038/modpathol.2017.175
Dong HY, Wilkes S, Yang H (2011) CD71 is selectively and ubiquitously expressed at high levels in erythroid precursors of all maturation stages. Am J Surg Pathol 35(5):723–732. https://doi.org/10.1097/pas.0b013e31821247a8
Wang W, Wang SA, Medeiros J, Khoury JD (2017) Pure erythroid leukemia. Am J Hematol 92:292–296. https://doi.org/10.1002/ajh.24626
Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD (2009) p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4(1):37–48. https://doi.org/10.1016/j.stem.2008.11.006
Montalban-Bravo G, Benton CB, Wang SA, Ravandi F, Kadia T, Cortes J, Daver N, Takahashi K, DiNardo C, Jabbour E, Borthakur G, Konopleva M, Pierce S, Bueso-Ramos C, Patel K, Kornblau S, Kantarjian H, Young KH, Garcia-Manero G, Andreeff M (2017) More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia. Blood 129(18):2584–2587
International Standing Committee on Human Cytogenetic Nomenclature (2020) An International System for Human Cytogenetic Nomenclature. In: McGowan-Jordan J, Hastings RJ, Moore S (eds) Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature, Karger, Basel, Switzerland
Weinberg OK, Hasserjian RP, Baraban E, Ok CY, Geyer JT, Philip JKSS, Kurzer JH, Rogers HJ, Nardi V, Stone RM, Garcia JS, Hsi ED, Bagg A, Wang SA, Orazi A, Arber DA (2019) Clinical, immunophenotypic, and genomic findings of acute undifferentiated leukemia and comparison to acute myeloid leukemia with minimal differentiation: a study from the bone marrow pathology group. Mod Pathol 32(9):1373–1385. https://doi.org/10.1038/s41379-019-0263-3
Wang SA, Tam W, Tsai AG, Arber DA, Hasserjian RP, Geyer JT, George TI, Czuchlewski DR, Foucar K, Rogers HJ, Hsi ED, Bryan Rea B, Bagg A, Dal Cin P, Zhao C, Kelley TW, Verstovsek S, Bueso-Ramos C, Orazi A (2016) Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol 29(8):854–864. https://doi.org/10.1038/modpathol.2016.75
Saft L, Karimi M, Ghaderi M, Matolcsy A, Mufti GJ, Kulasekararaj A, Göhring G, Giagounidis A, Selleslag D, Muus P, Sanz G, Mittelman M, Bowen D, Porwit A, Fu T, Backstrom J, Fenaux P, MacBeth KJ, Hellström-Lindberg E (2014) p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica 99(6):1041–1049. https://doi.org/10.3324/haematol.2013.098103
Bártek J, Bártková J, Vojtĕsek B, Stasková Z, Lukás J, Rejthar A, Kovarík J, Midgley CA, Gannon JV, Lane DP (1991) Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene 6(9):1699–1703
Brosh R, Rotter V (2009) When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 9(10):701–713. https://doi.org/10.1038/nrc2693
Pant V, Quintás-Cardama A, Lozano G (2012) The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood 120(26):5118–5127. https://doi.org/10.1182/blood-2012-05-356014
Code availability
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors have substantially contributed to conception and design, acquisition of data, or analysis and interpretation of data; drafted the article or revised it critically for important intellectual content; and have approved the final version to be submitted for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Mayo Clinic Institutional Review Board and we certify that the study was performed in accordance with appropriate ethical standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alexandres, C., Basha, B., King, R.L. et al. p53 immunohistochemistry discriminates between pure erythroid leukemia and reactive erythroid hyperplasia. J Hematopathol 14, 15–22 (2021). https://doi.org/10.1007/s12308-020-00431-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-020-00431-7