
Abstract
Messenger RNA (mRNA) recently emerged as an appealing alternative to treat and prevent diseases ranging from cancer and Alzheimer’s disease to COVID-19 with significant clinical outputs. The in vitro-transcribed mRNA has been engineered to mimic the structure of natural mRNA for vaccination, cancer immunotherapy and protein replacement therapy. In past decades, significant progress has been noticed in unveiling the molecular pathways of mRNA, controlling its translatability and stability, and its evolutionary defense mechanism. However, numerous unsolved structural, biological, and technical difficulties hamper the successful implementation of systemic delivery of mRNA for safer human consumption. Advances in designing and manufacturing mRNA and selecting innovative delivery vehicles are mandatory to address the unresolved issues and achieve the full potential of mRNA drugs. Despite the substantial efforts made to improve the intracellular delivery of mRNA drugs, challenges associated with diverse applications in different routes still exist. This study examines the current progress of mRNA therapeutics and advancements in designing biomaterials and delivery strategies, the existing translational challenges of clinical tractability and the prospects of overcoming any challenges related to mRNA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The growing interest in messenger RNA (mRNA) has resulted in a groundbreaking class of potent drugs to target “undruggable” genes and gene products, generating a new paradigm for managing various diseases from cancer to COVID-19. In cancer therapy, mRNA shows significant therapeutic prospects to restore or replace mutant genes with loss of functions in tumor cells. Restoring the expressions of undruggable cardinal genes through mRNA exerts significant growth suppressive action against cancers cells (Xiao et al. 2022). In addition, CRISPR/Cas9 mRNA also adds a new dimension to cancer gene therapy which facilitates the specific inactivation of overexpressed gene responsible for tumorigenesis through site-specific targeted genome editing (Zhang et al. 2022a, b, c). Messenger RNA, a natural biomolecule, is an intermediary for translating genetic information from genes into a functional protein. It is not a permanent genetic molecule nor a stable, functional product. Unique features of the mRNA, such as adaptability and its transient nature, give tremendous hope to patients as comprehensive therapeutic applications in vaccination, protein replacement therapies, and cancer treatment compared to most existing drugs (Hajj and Whitehead 2017). The recipients’ immune system can be quickly trained recognize and kill tumors with the help of mRNA. The study of mRNA began with its discovery in 1961 (Brenner et al. 1961), enabling the unveiling of mRNA’s structural and biological function, its molecular path of translation and metabolic pathways, and its role in protein synthesis. In the early 70 s, several groups attempted to deliver naked mRNA directly using different methods to translate it into a target protein in different cell lines (Lane et al. 1971; de Maeyer-Guignard et al. 1972; Furusawa et al. 1974; Loyter et al. 1975; Stacey and Allfrey 1976). Subsequently, in 1978, the first successful translation of rabbit globin mRNA into mouse spleen lymphocytes using unilamellar liposomes were reported (Dimitriadis 1978). Many unresolved issues remain despite successfully translating the isolated mRNA into a protein. Immunogenicity, biological instability, poor cellular internalization, and production costs slowed down mRNA therapeutics’ clinical application. In 1990, the first preclinical study of in vitro-transcribed (IVT) mRNA in a mouse model was reported as the first step of mRNA therapeutic development (Wolff et al. 1990). Five years later, the mRNA polynucleotide vaccine was developed and exerted a notable immune response against the tumor-associated carcinoembryonic antigen (CEA) and reduced tumor growth (Conry et al. 1995). Consequently, in 1999, mRNA-transfected dendritic cells (DCs) were brought into phase I clinical trials for renal and prostate cancer management (Gilboa and Vieweg 2004). Since then, mRNA therapeutics and innovative delivery methods and vehicles have shown tremendous potential for preventing and treating multiple types of cancer (Sahin et al. 2014; Uchida et al. 2016; Oberli et al. 2017).
Structurally, mRNA is composed of a 5’ cap structure, 5’ UTR (untranslated region), coding region, and 3’ UTR and a poly-A tail. The 5’ cap structure comprising 7-methyl guanosine (m7G) is involved in the initiation of translation. This element binds with the eukaryotic translation initiation factor 4E (EIF4E), nuclear mRNA export, and stabilization of mRNA by escaping from exonuclease degradation and innate immune attack (Sonenberg and Gingras 1998; Calero et al. 2002; Parent a Bisaillon 2006; Anderson et al. 2010; Li and Kiledjian 2010; Tavernier et al. 2011). The poly-A tail contains 100–250 adenosine residues at the 3’ end of mRNA in eukaryotic cells, responsible for expediting nuclear export, translation, and mRNA stability. It also impacts the splicing and post-transcriptional processes (Lewis et al. 1995; Wickens et al. 1997; Neugebauer 2002; Calvo and Manley 2003; Karikó et al. 2004; Proudfoot 2004; Goldstrohm and Wickens 2008; McIvor 2011; Andries et al. 2013). The untranslated regions at the 5’ and 3’ ends are the non-coding region of mRNAs that do not directly involve protein synthesis. It plays a vital role in mRNA stabilization, ribosome recruitment, starting of codon choice, and protein expression level (Mignone et al. 2002; Gebauer and Hentze 2004; Hinnebusch et al. 2016; Mayr 2017; Leppek et al. 2018; Xiong et al. 2018). The CAP and poly-A tail structures of in vitro transcribed (IVT) mRNA are tuned to facilitate the biological properties of mRNA therapeutics (Peng and Schoenberg 2005; Mockey et al. 2006; Grudzien-Nogalska et al. 2007; Kowalska et al. 2008, 2014; Jemielity et al. 2010; Kuhn et al. 2010). Since the discovery of mRNA, scientists have struggled to boost mRNA’s translatability and stability to maximize control over specific therapeutic applications.
To an extent, the augmented translation, adequate biological stability, and less immunogenic activity of mRNA are achieved by modifying the untranslated (UTRs) regions, cap structure, coding regions, and poly-A tail of the mRNA structure (Jemielity et al. 2010; Anderson et al. 2011; Strenkowska et al. 2016). Previous studies on structural modification of IVT-mRNA substituted uridine residues with N-methyl pseudouridine (TriLink). This element increased translation of encoded protein in the spleen at significantly low doses (0.015 mg/kg) and was less immunogenic in mouse models (Karikó et al. 2008). The modified mRNA could not trigger the Toll-like receptors (TLRs) (Karikó et al. 2005), lowering the secretion of IFN-α and thus displaying comparatively higher stability and suppression of immune responses than unmodified mRNA. In addition, naked mRNA in systemic circulation encountered several factors, such as RNase degradation, RES clearance, cellular internalization, and release of mRNA from delivery vehicles into target cell cytoplasm that limits the efficiency of mRNA drugs. Bypassing these extracellular and intracellular barriers of mRNA after systemic administration to reach the target cell cytoplasm remains the major obstacle, especially for broader clinical applications. In order to function in preclinical and clinical studies, naked mRNA therapeutics must be furnished with a delivery vehicle for valuable clinical output. Although many viral and non-viral carriers have been used to transport IVT-mRNA in clinical and preclinical studies (Zohra et al. 2009; Guan and Rosenecker 2017), ideal and safer delivery materials have not been identified. Therefore, areas of concern, such as biological half-life, specific delivery of mRNA, and its complex pharmacology, must be clarified to propel the systemic application of mRNA for fighting cancer.
With the fast-track development of engineered mRNA and mRNA vaccines to manage the SARS-CoV-2 outbreak in 2019, mRNA therapeutics is booming. In December 2020, the world witnessed the first FDA-approved mRNA drug, the Pfizer-BioNTech vaccine BNT162b2 after a record-breaking authorization time (Polack et al. 2020). The stunning success of nanoparticle-based mRNA vaccines gave new hope for mRNA nanotherapeutics development against cancer., biotechnology companies Biotechnology companies like Moderna, BioNTech, and CureVac have started developing mRNA cancer vaccines. More than 20 mRNA-based cancer vaccines have undergone clinical trials to manage different malignancies like melanoma, non-small cell lung cancer, and colorectal carcinoma (Zhu et al. 2022). For example, DCaT–RNA is undergoing the final stage of clinical trials (NCT01983748) (Wang et al. 2022). Thus, the advanced mRNA technology and massive data from manufacturing mRNA-based cancer vaccines and human recipients show promise for the application of mRNA for tumor treatment.
In this study, the researchers explore the various approaches in mRNA therapeutics development and the progress in mRNA delivery technologies. The anomalies of the commercial development of nanoparticles (NPs)-based mRNA drugs and the key challenges that impact the rapid succession in clinical trials are also highlighted.
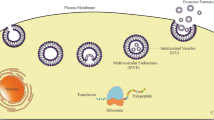
Based on Fig. 1, a linearized plasmid DNA template with target gene sequence is utilized to synthesize IVT-mRNA. The synthesized mRNA consists of CAP 5’ untranslated region (UTR), coding region, 3’ untranslated region (UTR) and the poly-(A) tail. All of the components of mRNA structure regulate the translation efficiency and stability of IVT-mRNA. This structure is coupled with transporting vehicles through electrostatic interaction to form nanocomplexes. After systemic delivery, mRNA-nanocomplexes accumulate in the target cell, bypassing extracellular barriers. The IVT-mRNA-nanocomplexes are internalized into the cell through endocytosis. The rate of cellular uptake can be modulated by modifying the targeting ligands. The new internalized mRNA facilitates endosomal escape by disrupting endosomes and releasing the mRNA into the cytosol. The released mRNA is transported to the ribosome, where the protein of interest can be translated. In vaccines, the antigen encoding of mRNA is transcribed into the desired antigen and activates the immune system.

Schematic representation of basic IVT-mRNA pharmacology
The basic concept of IVT-mRNA pharmacology
Extensive research on mRNA therapeutics enables a reasonably straightforward procedure for in vitro transcription in the laboratory. Therapeutic mRNA produced by the IVT method aims to synthesize specific functional proteins to halt the disease progression by propagating a robust immune response or improving the expression of a specific gene (Baptista et al. 2021). The mRNA synthesis starts from a DNA template (a linearized plasmid vector, PCR product and double-stranded oligonucleotide). The DNA template contains an RNA polymerase-specific promoter derived from T7, T3, or SP6 bacteriophages, with the intended sequence for the specific protein of interest (Fig. 1). The T7 or SP6 RNA polymerase traverses from the DNA template strand 3’ to 5’ end. It produces mRNA molecules in the 5’ to 3’ direction in the presence of ribonucleotide triphosphate (Pardi et al. 2013, 2018; Sahin et al. 2014; Weissman 2015;). In addition, a 7-methyl guanosine cap is incorporated into the IVT reaction with an enzyme for augmenting the translation and stability of mRNA. Finally, the DNA template is removed through DNase digestion and purified by conventional nucleic acid isolation methods, such as lithium chloride precipitation or column-based purification.
IVT mRNA can be delivered in-vivo, either naked or with delivery vehicles. Several approaches have been used to deliver naked synthetic mRNA, including (i) direct delivery via the intramuscular or intratracheal route, (ii) in vivo electroporation, and (iii) gene gun-triggered delivery (Fleeton et al. 2001; Kofler et al. 2004; Johansson et al. 2012; Mays et al. 2013; Cu et al. 2013; Zeyer et al. 2016). In addition, multiple delivery vehicles, including lipid NPs, polymeric NPs, and hybrid inorganic–organic NPs, enhance the delivery efficiency of mRNA. In general, two primary modalities are known to treat multiple types of cancer using IVT mRNA. The first is ex vivo delivery of IVT mRNA where blood cells are collected from the patient, isolated, and transfected in a cell culture dish to express a tumor-associated antigen into collected patient cells. These transfected cells are then returned to the patient to attack cancer cells. This method is widely used for T-cell- and dendritic cell (DC)-based cancer immunotherapy (Pardi et al. 2018; Mukalel et al. 2019). The other method is the systemic delivery of mRNA by intradermal, intravenous or intramuscular routes. This method increases the expression of cancer-limiting genes. Upon IVT-mRNA administration, the substance will face a series of hurdles in systemic circulation. With high-density negative surface charges and a size three to four times larger than the molecules capable of diffusing across the cell membrane (Yin et al. 2014; McNamara et al. 2015; Ramamoorth and Narvekar 2015; Kowalski et al. 2019). Single-stranded mRNAs are policed by a mononuclear phagocyte system (MPS) and repulsed by a negatively charged cell membrane, resulting in, respectively, pre-mature degradation and poor cellular uptake with a rate of less than 1 in 1000 molecules (Sahin et al. 2014).
Several delivery vehicles have improved the stability in systemic circulation and facilitate mRNA cellular penetration. After extravasation from the extracellular space, IVT mRNA must be released from delivery vehicles into the cell cytoplasm, the initial pharmacodynamics compartment of IVT mRNA. The intercalation of vectors into endosomal membranes through membrane disruption or membrane permeability releases therapeutic mRNA (Vaidyanathan et al. 2016a, b). The released mRNA is subsequently transported to the ribosome, a tiny protein-synthesizing machine and binds the ribosomal complex. Translation begins with the scanning of the mRNA sequence from the 5’ to 3’ direction by the ribosomal complex (Fig. 1). This process is followed with the recognition of the start codon where transfer RNA (tRNA) brings amino acids for pairing and linking together to elongate the peptide chain until a termination codon is reached. The synthesized protein undergoes post-transcriptional modifications and is translocated to the desired cell compartment within the host cell or neighboring/distant cellular organs to modulate target gene expression.
Benefits and shortcomings of using mRNAs vaccines and therapeutics
The remarkable advancement in mRNA technology facilitates clinical opportunities for mRNA therapeutics in vaccination, protein replacement therapy, regenerative medicine, cellular reprogramming, and genomic editing. mRNA has several unique features appealing to researchers. First, it does not integrate into the host cell’s genome, a common problem in viral-based vaccines and DNA therapies. Therefore, there is no risk of insertional mutagenesis. Second, mRNA does not need to enter the nucleus to function at an optimal level since it gets translated into the cytosol, other DNA therapies which require nuclear envelope breakdown during cell division to access the nucleus (Sahin et al. 2014). Third, mRNA has a transient effect on cells. Normal physiological pathways destroy it once it has served its purpose, allowing better control over pharmacokinetics and dosage (Petsch et al. 2012). Finally, it reduces the risk of inducing tolerance (Pollard et al. 2013). Since the proteins are manufactured in the host cells, there are no protein aggregations or impurities from cell culture except for mRNA manufacture (Sahin et al. 2014). The manufacturing process of mRNA is inexpensive, with a high reaction yield. It also has a better safety profile, as the manufacturing process requires no toxic reagents and involves only cellular or animal components. The ability of mRNA to be manufactured and scaled rapidly makes it an ideal tool for tackling potential pandemics (Bahl et al. 2017). Protein replacement therapies can avoid the cold chain logistics associated with protein products since the mRNA can be stored in lyophilized form (Kwon et al. 2018). In particular, large-scale cell-based protein production through traditional recombinant technology has some intrinsic drawbacks like manufacturing facilities, quality control, and cold chain infrastructure for transportation and long-term storage. These shortcomings of protein products employed for protein replacement therapy can be easily overcome by utilizing messenger RNA therapeutics. mRNA has additional features specific to regenerative medicine, for example, engineering stem cells by inducing the production of homing proteins. Since mRNA expression is transient, this provides an additional advantage over DNA treatment or surface modification with proteins which could have side effects due to the permanence of the procedure and disruption of the cell surface membrane (Kauffman et al. 2016). mRNA induces pluripotency and avoids residual expression of transgenes encountered with viral vectors from insertional mutagenesis. The transient activity of mRNA also makes it suitable for gene editing without undesirable and prolonged expression of nucleases due to the risk of nonspecific editing (Sahin et al. 2014). Despite the advantages of using mRNA to develop vaccines and therapeutics, several limitations must be addressed to improve the effectiveness of these treatments. First, the volume of RNA needed to induce a therapeutic outcome, particularly in protein replacement therapy, is difficult to determine (Sahin et al. 2014). Second, IVT mRNA can stimulate the immune system and function as an intrinsic adjuvant. This feature is excellent for mRNA vaccines, although undesirable for protein replacement applications.
As for vaccines, IVT mRNA only stimulates CD8 + T cells through cell-mediated immunity without activating CD4 + cells, leading to a less effective immune response. However, a recent study shows that the mRNA vaccine can stimulate CD4 + T cells (Painter et al. 2021). It is also challenging to find an adjuvant that will not interfere with mRNA uptake (Pollard et al. 2013). mRNA has a short half-life in vivo due to nucleases in the blood and pattern recognition receptors. It is also more sensitive to oxidation than DNA (Brito et al. 2015). Therefore, many mRNA treatments require a delivery system. Translation of mRNA treatments to clinical use has been challenging, with clinical trials showing modest results compared to expectations from preclinical testing (Pollard et al. 2013; Pardi et al. 2018). Further, there are still supply limitations to consider. While all the supplies needed for mRNA manufacturing are obtainable at GMP standards, some are only available at a limited scale or high cost (Pardi et al. 2018).
Commonly used delivery vehicles are lipids, polymers, peptides, and hybrid inorganic and organic nanoparticles. The mRNA lies at the core of the nanoparticles or adsorbed onto the surface of carriers surrounded by lipid shells. Negatively charged mRNA forms complex nanocarriers through electrostatic interaction.
Design of nanocarriers for vectoring mRNA therapeutics
Purpose of transporting vehicles
Nanomedicine has revolutionized the management of many diseases like cancer. The benefits include improved pharmacodynamics, pharmacokinetic and safer toxicity profiles. Nanoparticles (NPs) ranging from 1 to 100 nm are assembled with several therapeutics like small molecular drugs, nucleic acids, proteins, and peptides for sustained and targeted drug delivery (Karim et al. 2017, 2018; Ramasamy et al. 2017, 2019; Ruttala et al. 2018; Jahan et al. 2021). Additionally, theranostic NPs added a new feature and are being extensively studied for traceable drug delivery systems, molecular imaging, and tracking intracellular processes (Thangam et al. 2022; Zhang et al. 2022a, b, c). Structurally NPs possess uniform smaller sizes, large surface-to-volume ratios, and tunable surface functionalization properties. This feature offers less uneven bio-distribution, improves encapsulation of more drugs and nucleic acids, and facilitates controlled drug release and efficient intracellular delivery. However, the rate of successful translation from bench to bedside remains very low despite an increasing number of preclinical studies. Several biological, pharmaceutical, and translational barriers, such as the design of scalable transporters, lack of efficacy in human trials, and clinical safety profiles are considered the root causes of failure in clinical trials. Therefore, mRNA therapeutics is a promising alternative to plasmid DNA-based gene therapy.
Further, mRNA is large (103–105 nucleotides), negatively charged, hydrophilic in nature, and susceptible to nuclease-mediated degradation, resulting in a short blood half-life and limiting the cellular entry of mRNA into the target cell. In order to navigate these barriers, it is obligatory to equip the mRNA with a transporter to protect it from enzymatic degradation in the systemic circulation, avoid immune detection, and lessen the non-specific interactions with serum proteins. It also helps bypass the cell membrane and release the payload from delivery vehicles for subsequent translation without activating any immune response. Recent studies indicate a wide variety of NPs, including lipid and lipid derivatives, polymers, proteins, inorganic materials, and hybrid particles for the delivery of mRNA (Fig. 2). These NPs show favorable pharmacokinetics and potent pharmacological effects against multiple chronic diseases (Ramasamy et al. 2021). The composition and selection of NPs for mRNA therapeutics ensure stability and transfection efficiency. mRNA triggers endosomal escape, prevents immune activation and provides clinically translatable NPs-based mRNA drugs.

Major delivery platforms for IVT-mRNA transportation
Liposomes
Among the non-viral vectors, liposomes are the mainstay of IVT-mRNA delivery. Liposomes are spherical vesicles comprising monolamellar or multilamellar phospholipid bilayers enclosing an aqueous core to house therapeutics of interest. Preliminary, cationic lipids, such as DOTMA (1,2-di-O-octadecyl-3-trimethylammonium-propane), DOTAP (1,2-dioleoyl-3-trimethylammonium-propane) and zwitterionic DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) that formed lipoplexes with IVT-mRNA through electrostatic interactions used alone or in combination with other materials to deliver the mRNA (Torchilin 2005). In preclinical studies, cationic liposomes showed comparatively low transfection efficiency compared to their viral counterparts. Its efficacy, stability, and transfection efficiency improved through condensed protamine and mRNA into nano-sized conjugates. This feature protects against biological degradation (Palamà et al. 2015a, b). Men et al. (2018) attempted delivery of vesicular stomatitis virus matrix protein (VSVMP) encoding mRNA with liposome-protamine complex to induce apoptosis in colon cancer cells. The liposome prepared from DOTAP and cholesterol (1:1, mol/mol) complexed with protamine cationic liposome–protamine complex (CLPP) for improved stability in systemic circulation and efficient delivery of mRNA into the target site showed a higher expression level of reporter gene EGFP (Enhanced green fluorescent protein) after 24 h of transfection in 293t and C26 cells.
In the C26 colon cancer mouse model, the CLPP/VSVMP mRNA complex treated mouse showed an elevated level of VSVMP mRNA with no severe metastases in the abdominal cavity and a notable reduction of tumor growth. The structure had a 61.6% growth inhibition rate against C26 colon cancer cells compared to the control group. The intratumoral injection of CLPP/VSVMP mRNA triggered apoptosis and angiogenesis inhibition without any toxicity (Kauffman et al. 2016; Men et al. 2018; Palza et al. 2019).
Similarly, Zhang et al. (2019a, b, c) used liposome-protamine complex (CLPP) coupled with mRNA encoding survivin-T34A gene to evaluate and compare the anticancer efficacy with plasmid DNA counterpart against colon cancer. The CLPP bound with mRNA through electronic interaction and condensed to nano-sized particles with a particle diameter of 186.1 ± 3.1 nm exerted considerable transfection efficiencies of 24.6 ± 1.5% and 40.3 ± 1.6% after 24 and 48 h of post-transfection in C26 cells. Enhanced cytotoxic effects were found, with an inhibition rate beyond 58% compared to the plasmid DNA treated group. The metastasis and tumor regression studies of CLPP/mSur-T34A, complex in a subcutaneous C26 colon carcinoma model showed lesser metastasis in the abdominal and pulmonary regions. The structures augmented antitumor efficacies after local and intravenous administration compared to plasmid DNA without any changes in major organs (Zhang et al. 2019a, b, c).
Apart from protamine, several other transfection agents have been deployed to improve the transfection efficiency of the liposome-mRNA complex. Recent modified liposome-mediated mRNA delivery progressed tremendously in antigen-specific vaccines for cancer immunotherapy due to higher transfection efficiency and self-adjuvant effects. Such successful exploration led to commercially viable lipid-based transfection agents, including Lipofectamine (Invitrogen) and Trans-IT (Mirus), for the delivery of mRNA in vitro and in vivo (Hajj and Whitehead 2017). These modifications increase biological stability and translation efficiency, reduces mRNA’s immunogenicity and enable the full potential of antigen-specific mRNA vaccines. Although liposome-based transfecting agents showed promising transfection efficacy in vitro, they exerted significant toxicity, such as liver damage and inflammatory responses in animal model studies (Karikó et al. 2008).
Arya et al. (2020) introduced another cationic liposome-based transfecting agent InstantFECT. The structure delivered pseudouridine-modified mRNA encoding the melanoma-associated antigen with a simple and translatable protocol. Further, the mRNA condensed into cationic liposome-based transfecting agents InstantFECT through electrostatic interactions (Arya et al. 2020). The results indicate a higher expression of GFP with a transfection efficiency of 37–42% in HEK cells observed beyond 96 h of the luciferase mRNA InstantFECT nanocomplex injection. This finding is the highest luciferase activity compared to the application of commercial liposome and Trans-IT (Mirus) in animal model studies. Highly penetrable InstantFECT nanocomplex with modified OVA mRNA showed remarkable regression of tumor and long-term prophylactic action of mice against B16-OVA melanoma, indicating a strong T cell response against OVA through maturing dendritic cells. The long survival rate and no toxicity of mice receiving therapeutic and prophylactic pseudouridine-modified mRNA-InstantFECT nanocomplex represent a wide range of safety profiles in animal model studies (Arya et al. 2020).
Dendritic cells (DCs) are the principal elements in instigating, processing and regulating antigen specific immune responses regarding a state of activation or tolerance. DCs are the most robust antigen-presenting cells that mediate antitumor responses through the uptake of tumor-derived particles and activation of CD8 + T cell responses (Nagy et al. 2021; Sasaki et al. 2022). These unique features of DCs extensively studied for the clinical application of mRNA cancer vaccines places them at the forefront of the cancer management. Despite the excellent immune response of liposomes-based neoantigen-specific mRNA vaccines in many clinical studies, the delivery efficiency and the immune response of dendritic cells (DCs) vaccines must be maximized (Zhang et al. 2018, 2020a, b). The author found biocompatible cholesterol-modified antimicrobial peptide-based immune adjuvant DP7 to boost the immune responses of mRNA-based vaccines. The DOTAP/DP7-C increased the transfection efficiency of encoded mRNA into DCs and facilitated the expression of eGFP mRNA into 293 T cells. The higher penetrating efficiency of DP7-C allowed more liposome-loaded neoantigen mRNA into DCs through caveolin and clathrin-dependent pathways (Zhang et al. 2018). This finding resulted in significant antitumor effects and observable preventive anti-proliferative actions in an animal model study. The dual action of DP7-C mediates improved mRNA delivery and DC activation by TLRs signaling pathways. Therefore, antigen-specific cytotoxic T lymphocyte reactions induced a higher immune response and elicited a superior antitumor efficacy by DOTAP/DP7-C/ neoantigen mRNA DC vaccine (Zhang et al. 2020a, b).
The delivery of mRNA biologics for correcting the altered protein function in the brain is a potential paradigm to mitigate the progression of neurological disorders. The most common delivery methods and technologies for mRNA transport, i.e., ex-vivo delivery and systemic route, are more challenging and strenuous, expensive in synthesis, with bypass harsh biological barriers. Moreover, specific intranasal delivery of mRNA to the brain is required as systemic delivery results in lower therapeutic doses (Jogani et al. 2007; Sousa et al. 2019). mRNA therapeutics for Alzheimer’s, Parkinson’s, and frontotemporal lobar degeneration (FTLD) have not been established (Wolfe 2014; Singh et al. 2019). However, Dhaliwal et al. (2020) successfully delivered protein therapeutics into the brain using nontoxic liposomes (GDNF) (Migliore et al. 2010, 2014) or liposomes in the gel system (BDNF AntagoNAT Oligonucleotides) (Pawar et al. 2018). Later, the authors developed cationic liposome-based mRNA complexes delivered through the nasal route for brain-specific delivery (Dhaliwal et al. 2020). The intranasal delivery of GFP-mRNA/Liposomes transfers mRNA into the brain’s trigeminal nerve pathways (Yadav et al. 2015). It bypasses the blood–brain barrier (BBB) and hepatic first-pass metabolism. This process results in a unique GFP-mRNA expression in the brain up to 24 h with minimal systemic exposure (Yadav et al. 2015).
Lipid nanoparticles (LNPs)
Though cationic lipids are promising in mRNA delivery, they have shortcomings, such as instability in systemic circulation and immunogenicity. For example, intravenously administered cationic liposomes cause liver damage, exert strong interferon-γ triggered inflammatory responses, and are neutralized by serum anionic opsonin proteins (Cherng et al. 1996; Uzgün et al. 2011). Therefore, cationic lipids must be redeveloped in lipid nanoparticles (LNPs) to increase stability, biocompatibility, and transfection efficiency. Generally, LNPs comprise ionizable lipids, cationic lipids, or polymeric materials. This structure releases the cargo inside the cytoplasm through the proton sponge effect (Huotari and Helenius 2011). LNPs include zwitterionic lipids (e.g., 1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine [DOPE]), cholesterol, and PEG. Helper lipids (DOPE) comprise lipid resemble and cell membranes. These structures promote fusion with the cell membrane, accelerating cellular internalization and endosomal release. Simultaneously, cholesterol fills the gap between lipids within the liposomal membrane to stabilize the LNP (Li and Szoka 2007). To increase systemic stability, PEG reduces opsonization by serum opsonins and reticuloendothelial system (RES) clearance.. Further, the efficacy of LNPs relies on the ratio of ionizable lipids: helper lipids: cholesterol, and PEG constituents. Pardi et. al (2015) evaluated the kinetics and pattern of Luc-mRNA expression upon intravenous, and other injection doses of 0.005–0.250 mg/kg nucleoside-modified mRNA conjugating LNPs. Mice receiving 5.0 µg of mRNA-LNPs complexes via the intravenous route showed the highest translation of encoded protein in the liver, represented by the strongest bioluminescent signal and amount of protein produced up to 24 h post-injection (Pardi et al. 2015). However, intramuscular and intradermal injections prolonged mice’s active translation of encoded protein. The variation in expression pattern, duration of protein translation, and the amount of produced protein of mRNA-LNP complexes at different routes turn on the dose, available potential target cells at the injection site, trafficking of mRNA therapeutics to the target cells, cellular uptake pattern, and turnover rate of intended protein into the site of action.
In addition, extensive research identified new LNP mRNA carriers by studying the structure–activity relationship of existing LNPs to maximize the delivery of mRNA in different human conditions (Pardi et al. 2015). Classical orthogonal experimental design has been utilized to identify critical determinants and the effects of transformation within the parent structure on delivery efficiency. The magnitude and efficiency of LLN (lipid-like nanoparticles) based mRNA transfection improved by modifying the core structure, linkers, and lipid chain length (Love et al. 2010; Dong et al. 2014). Li et al. (2015) observed a new derivative of N1, N3, N5–tris (2-aminoethyl) benzene-1, 3, 5-tri carboxamide (TT) derived LLNs influenced by their previous success on the delivery of siRNA in animal models (Zhang et al. 2013; Li et al. 2015). A classical orthogonal array assay was performed to select the best formulations from the possible 256 yields based on their physicochemical properties, entrapment efficiency, and transfection efficiency. The higher delivery efficiency of 350 times optimized TT3 LLNs is attributed to higher transfection of Luc-mRNA in a human hepatoma cell line (Hep3B cells). The level of hFIX (a blood clotting factor) protein reinstated to its average physiological value after intravenous injection of optimized O-TT3 LLNs conjugated mRNA encoding human factor IX (hFIX) in FIX-knockout mice (Li et al. 2015). In another study, the authors found that rearrangement in structural conformation in the main core of LLNs resulted in improved stability and more efficient mRNA delivery to the target cell. By studying the structure–activity relationship (SAR) of LLNs and a new optimized O-TNT-b10, LLNs have been developed by exchanging hydroxyl and amino groups and modifying carbon chain length, which increased mRNA delivery efficiency ten times more than unmodified LLNs. The lead LLNs in mice showed significant translation and expression of luciferase in the liver and spleen. This finding is without any histopathological changes in the morphology of significant organs (Li et al. 2016). In addition, the therapeutic efficacy of LNP-based mRNA can be improved by a combinational therapy with chemotherapy drugs against aggressive therapeutic targets lacking Triple-negative breast cancer (TNBC). For example, Zhang et al. (2019a, b, c) combined classical chemotherapeutic drugs paclitaxel with amino lipid nanoparticles (PAL) through an ester bond and p53 mRNA to form PAL P53 mRNA NPs. The sorted analogs of amino lipids exerted better stability, encapsulation efficiency, and release profile of installed paclitaxel. The paclitaxel and p53 mRNA interacted with antitumor action in an orthotopic TNBC model with an extended median survival time (Zhang et al. 2019a, b, c).
On the other hand, in cancer immunotherapy, the induction of a robust cytotoxic T cell response through the synthesis of intracellular antigens is the crucial determinant factor in exploiting the immune system for cancer management. Several research groups targeted specifically expressed tumor-associated antigens to regulate cytotoxic T Cells (CTLs) or cluster of differentiation 8 (CD 8) T cells responses (Melief et al. 2015; Palucka & Coussens 2016) to reduce the tumor burden. Subcutaneous administration of B-11 conjugated mRNA encoding OVA, tumor-associated antigens gp100 and TRP2 showed a potent immune response, and heightened CD 8 T cells’ proliferation caused significant antitumor action in the mice model. Accordingly, the effects of individual components and the ratio of lipid nanoparticles on mRNA transfection, CD 8 T cell level and inflammatory responses optimized LNP formulations B-11 in the delivery of mRNA vaccines (Oberli et al. 2017). Overall, remarkable survival was achieved by overcoming self-tolerance in a transgenic mouse melanoma model (Oberli et al. 2017).
mRNA endosomal escape after endocytosis into the target tumor cell cytoplasm limits successful mRNA transfection. It is strongly desirable to release the conjugated mRNA at the early endosomal stage to get quick therapeutic responses and prevent drug loss inside the cell cytoplasm. Previous studies applied different pH-dependent cleavable agents or pH-responsive nanomaterials. These structures accelerate endosomal membrane disruption or destabilization in an acidic pH (Karim et al. 2019). Further, structural motifs of LNP amino lipids determined their stability, cellular permeability, endosomal release and tolerance (Sabnis et al. 2018). New LNP particles were optimized with a rational chemistry approach by replacing the linoleic tail with a primary ester-containing lipid tail. Lipid 5-based LNPs increased the fusogenicity and promoted endosomal maturation and destabilization. The improved mRNA cytosol achieved efficient, extended luciferase mRNA and human erythropoietin (hEPO) expressions. The translational feasibility of the optimized mRNA system for clinical trials was examined on non-human primates. Favorable pharmacokinetics without elevation of immunological markers existed in Cynomolgus monkeys (Sabnis et al. 2018).
Polymer nanoparticles
Cationic polymer condenses the negatively charged mRNA into nanoplexes and shuttles across the target cellular or subcellular membrane. In the past, cationic polymers have successfully transported nucleic acids. Their application as mRNA vehicles is also on the rise. Compared to lipid vectors, polymers are not as clinically advanced. However, their chemical diversity, structural flexibility, and scalable synthesis procedure make them appealing mRNA carriers in protein replacement and vaccine applications. Linear polyethylenimine (l-PEI) and poly-N,Ndimethylaminoethylmethacrylate P(DMAEMA) were also observed. The composition constituted copolymer oligo (ethylene glycol), methyl ether methacrylate (OEGMA), and N,N-dimethyl aminoethyl methacrylate (DMAEMA). Strong pDNA and siRNA delivery capabilities existed (Cherng et al. 1996; Uzgün et al. 2010). A lower ratio of N/P = 10 of linear polyethyleneimine (l-PEI) produces a comparatively highest expression of Luc-mRNA on bronchial epithelial cells (Uzgün et al. 2011).
However, to some extent, poly (ethyleneimine) (PEI) is ineffective in the delivery of mRNA therapeutics (Debus et al. 2010; Rejman et al. 2010). Triblock copolymers (DPE1) make cationic dimethyl aminoethyl methacrylate (DMAEMA) the core for mRNA binding. Systemic stability and quick endosomal mRNA release also improve (Cheng et al. 2012). The lead DPE1 applied reversible addition-fragmentation chain transfer (RAFT) polymerization that can transfect GFP in two immune cell lines, RAW 264.7 macrophages (77%) and DC2.4 dendritic cells (50%), along with an elevated level of antigen-specific T cells activation (Cheng et al. 2012). A series of polymer brush nanoparticles composed of amino and hydroxyl groups incorporated polymer backbone with alkyl brushes at the tail (Dong et al. 2016). The structure–activity relationship of polymer brush materials showed TarN3C10 with a higher amino group (N3), the tartrate sugar, and fewer alkyl tails (C10) modulate the delivery efficiency of mRNA. Moreover, intravenous administration of TarN3C10 conjugated erythropoietin (EPO) mRNA at a dose of 0.3 mg/kg resulted in a 1000 times higher EPO expression. It also induced luciferase expression in the liver and spleen (1000 fold) with a well-tolerated safety profile in mice (Dong et al. 2016).
Alternatively, PCL nanoparticles increase the colloidal stability in the systemic circulation, cellular internalization, and endosomal release (Bhavsar and Amiji 2008). For example, the FDA-approved polymer poly (ε-caprolactone) (PCL) nanoparticles that deliver GFP mRNA-protamine complex showed a significant GFP expression in NIH 3T3 fibroblasts, HeLa cells and MG63 osteoblasts cells. The PCL NPs synthesized by the emulsion-diffusion-evaporation method with a diameter of 247.43 nm had a higher loading efficiency and stability from their hydrophilic PCL core and PVA composite shell. Moreover, arginine-rich peptide protamine protects mRNA degradation inside the cell cytoplasm. The buffering capacity of multicationic protamine triggers mRNA release that mediates prolonged expressions of GFP in different cell lines (Palamà et al. 2015a, b). Other PCL nanoparticles for mRNA delivery include poly (β-amino ester) co-poly (caprolactone) terpolymers. Poly (β-amino ester)s (PBAEs) were screened from a library based on physicochemical properties and delivery efficiency (Anderson et al. 2003; Sunshine et al. 2012). The authors applied a premixing protocol to construct a PCL-based PBAE-mRNA complex. Three formulations of C1 intravenous injection doses (0.5, 0.25, and 0.125 mg kg−1 mRNA) were administered. The novel PCL-based PBAE had materials with controlled properties and tolerability. The structure introduced efficient luciferase mRNA in mice spleen without elevated liver enzymes (Capasso Palmiero 2018).
Besides adequate stability of polymeric nanoparticles-mRNA complexes in blood, selective delivery to target cell cytoplasm could be another vital issue. The targeted delivery may reduce the off-distribution-triggered systemic toxicity drug loss, thereby lowering the overall treatment cost (Jahan et al. 2021). Cancer cells often increase with overexpression of some specific receptors as targets for ligand-decorated targeted delivery. Chen et al. (2017) reported cRGD ligand decorated multilayered polymeric nanoparticles cRGD-PEG/PNIPAM-PLys (SH) for selective delivery of mRNA to target tumor cells through systemic route. PLys crosslinked with mRNA in a redox-responsive disulfide linkage to fabricate the nanosized structure. Thermo-responsive PNIPAM (poly (N-isopropyl acrylamide)) translocated palisade moiety to the shell. This action formed an intermediate barrier between PLys and PEG at physiological temperature to boost protection against enzymatic degradation. The intravenous injection of cRGD-PEG/PNIPAM-PLys(SH) encoded GFP (10 mg in 200 mL of 10 mM HEPES containing 150 mM NaCl) in aVb3 and aVb5 integrins overexpressed U87 tumors in mice. Exerted extended half-lives in the bloodstream augmented tumor deposition and an observable higher GFP expression than other experimental groups (Chen et al. 2017).
Recently, a group of researchers engineered cationic poly (β-amino ester) (PbAE) polymers functionalized with Di-mannose moieties via polyglutamic acid (PGA) linker to target macrophages. Several M1-polarizing transcription factors, interferon regulatory factor 5 (IRF5), and activating kinase (IKKβ) encoded mRNA associated NPs were delivered to different mouse models. This process causes the reverse transition of M2 (pro-tumoral) to M1 (antitumoral) in Tumor-associated macrophages (TAMs) cells for antitumor immunity (Zhang et al. 2019a, b, c). Infusions of Di-mannose coated mRNAs encoding IRF5/IKKβ based NPs were made in three animal models; advanced-stage ovarian cancer, metastatic melanoma, and glioblastoma. The results indicate improvised macrophage targeting and lower M2-like macrophages in tumor lesions. This finding significantly delays tumor progression by boosting immunosuppressive action. A successful anti-cancer strategy, either alone or in combination with current immunotherapies, would be a potential anticancer paradigm in cancer management (Zhang et al. 2019a, b, c). The combinatorial testing of 480 modified functional polyesters involved amine-A17-modified polyester nanoparticles with higher translatability and stability. This process is based on the transfection efficiency of luciferase mRNA in IGROV1 ovarian cancer cells (Yan et al. 2017). Furthermore, the optimized polyester particles were functionalized with 5% triblock copolymer PEO101 − PPO56 − PEO101 (F127) to improve biological stability by escaping protein corona formation onto the surface of nanoparticles. Their systemic administration (20 μg Luc mRNA) induced 30-fold higher luciferase transfection in mouse lungs, proving the credibility of 5% F127 for specific mRNA delivery to the lungs (Yan et al. 2017).
Hybrid NPs
With substantial success in preclinical and clinical studies, liposomes and polymeric NPs are the first-line nanocarriers for mRNA delivery. Despite their success, they have shortcomings like toxicity, instability in a harsh biological environment, and endosomal escape inside the cell. Several groups have proposed a hybrid composite of lipid, polymer, inorganic nanoparticles, or peptide molecules to overcome the challenges by exploiting the benefits of an individual carrier. The developed hybrid nanoparticles reduce the toxicity of existing vectors and increase the transfection efficiency of the desired protein. For example, a combined system of PEGylated cationic polymer histidylated polylysine and L-histidine-(N,N-di-n-hexadecylamine) ethylamide cationic liposomes (lipopolyplexes) delivers human melanoma MART1 (MelanA) antigen and significantly delays progression of B16 melanoma after systemic administration in a mouse. The synergic pH-dependent release effects of histidylated cationic polymer and lipids promote the rapid release of encoded mRNA inside the acidic compartment of early endosomes. This process produces adequate cytotoxic T lymphocytes (CTLs) for antitumor immunity (Mockey et al. 2007). The authors modified PEGylated cationic histidylated polylysine and L-histidine-(N,N-di-n-hexadecylamine) ethylamide cationic liposomes (lipopolyplexes) with 11 mol % β-D-mannopyranosyl-N-dodecylhexadecanamide (Man-lipid) injections in the liposome formulation. This process produced mannosylated liposomes (Man11-Lip100) to induce DCs transfection and therapeutic potency of MART-1 mRNA-loaded vaccines. The degree and magnitudes of splenic DCs (CD11c + cells) expressing EGFP triggered by EGFP Man11-LPR100 were four times greater than non-mannosylated liposomes after intravenous administration. The mannose moiety could improve the liposomal stability in the systemic circulation and mannose receptor-mediated augmented cellular entry resulted in increased DCs expression and antitumor immunity obtained by intravenous injection of MART-1 Man11-LPR100 (Perche et al. 2011).
Zohra et al. (2007; 2009) studied the core mechanism and significance of hybrid organic and inorganic nanoparticles and improved gene delivery with high expression efficiency. They developed the world’s first pH-responsive inorganic carbonate apatite nanoparticles. Carbonate apatite coated with DOTAP-mRNA complexes produced hybrid inorganic–organic nanomaterials. This structure resolves cationic liposomes deficiencies. Endocytosis occurs in the acidic environment of the endosome and releases mRNA through endosomal destabilization. The proton sponge effect has efficient and comparatively higher transfection of Luc-mRNA than pDNA (Zohra et al. 2007; 2009). The authors also combined the surface of DOTAP-Carbonate apatite complexes with fibronectin to enhance specific targeting and overall transgene expression in mammalian cells. The addition of fibronectin shifted the surface charge from positive to slightly negative. The movement reduces non-specific interaction with serum proteins in the bloodstream by lowering the non-specific bio-distribution of nanoparticles inside the body. Fibronectin coated DOTAP-carbonate apatite complexes binds integrin receptors overexpressed in cancer cells. The structure allows more cellular entry and exerts superior expression of Luc-mRNA (almost two-fold higher than non-functionalized apatite complexes) (Zohra et al. 2012).
Other authors tested pH-responsive biodegradable poly (β-amino ester) (PBAE) enveloped with a phospholipid bilayer (lipid-enveloped poly-1) NPs for cytosolic delivery of mRNA-based vaccines (Su et al. 2011). Introducing a lipid shell to the polycationic core increased the surface stability and reduced the toxicity of cationic polymer by protecting it from serum interactions. Intranasal delivery of lipid-enveloped poly-1 NP-encoded luciferase mRNA offered the best duration effect. Elicited consistently and with stronger luminescence signal, the effects were maintained even 12 h after post-injection compared to naked mRNA. It has been hypothesized that lipopolyplexes extend the biological half-lives. The charged polymeric backbone underwent acidification and increased osmotic pressure disrupting the endosomal membrane. The rupture of endosomal membrane at an early stage resulted in a quicker release of adequate mRNA in the target cell cytoplasm. This process improved the overall transfection efficiency of the encoded protein (Su et al. 2011). Another variant of pH-responsive hybrid nanoparticles, Oligoalkylamine-based cationic polymers, and lipids complexes have been used for systemic delivery of mRNA (Jarzębińska et al. 2016). A lead C12-(2-3-2) was selected from a small library generated by tuning an alkyl chain and inserting different chain length methylene groups between amines having superior buffering capacity at acidic pH and adequate stability. Systemic delivery of the most effective C12-(2-3-2) carrying mRNA-FLuc mediated potent protein expression in the liver of mice, peaking six hours post-injection (Jarzębińska et al. 2016).
In addition, a series of nanomicelles variants PAsp (DET), PAsp (TEP), and PAsp (TET) were synthesized by inserting two, three, or four repeats of aminoethylene units in the side chain of block copolymers N-substituted polyaspartamides. The combination of in PEG-PAsp(TEP)-Chol/mRNA nanomicelles produced a hydrophobic layer around nanomicelles-mRNA complexes, and showed robust stability against serum protein anionic macromolecules in blood. Systemic administration of mRNA encoding fms-like tyrosine kinase 1 (sFlt-1) with PEG-PAsp(TEP)-Chol reduced notable vascular density in a mouse model of pancreatic adenocarcinoma (Uchida et al. 2016). The kinetics of mRNA transfection in phagocytic and non-phagocytic cells by hybrid nanoparticles (LPNs) comprised poly (lactid-co-glycolid) (PLGA) core surrounded by cationic lipid 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA). Live-cell video microscopy analysis of DC2.4 cells treated with hybrid nanoparticles (LPNs) indicated that after 15 min of incubation, the NPs associated with the cell surface. The protein translation started from one to four hours. Compared with cationic polymer chitosan coating PLGA, hybrid nanoparticles (LPNs) showed higher transfected DC.2(80%) and A549 cells (60%). This finding is due to higher stability and adequate release of encoded mRNA in the cytoplasmic compartment (Yasar et al. 2018).
In a recent study, enhanced cancer vaccine potency combined different adjuvants with mRNA vaccines. The combined effects of activating immune response are highly effective and produce a more robust antitumor response than a single counterpart. Yang et al. (2019) unified adjuvants TLR 7 ligands (gardiquimod) with antigen-encoding mRNA for efficient stimulation and maturation of cytotoxic T cells. Hybrid nanomaterials of poly (lactic-co-glycolic acid) (PLGA)-backbone with lipid-shell were constructed to facilitate the packaging and delivery of hydrophobic nonlipid adjuvants gardiquimod into the system. Adjuvants loaded hybrid nanoparticles efficiently produced EGFP protein (29.05 ± 1.39%), OVA ovalbumin (OVA)-derived MHC I-restricted peptide (SIINFEKL) in DC cells with a higher level of DCs maturation markers (CD80, CD86, and CD40). In an animal model, adjuvants- hybrid nanoparticles exhibited intensified luciferase expression in the mouse spleen. Further, the results showed a more robust OVA-specific CD4 + T cell immune response and significant tumor growth delay in therapeutic and protective models (Yang et al. 2019). On the other hand, αGalCer, also known as KRN7000, is an invariant natural killer T (iNKT) adjuvants co-delivered with mRNA encoding for tyrosinase-related protein 2 (TRP2). A hybrid lipopolyplex poly-(β-amino ester) polymer (PbAE) core entrapped into a lipid shell composed of multivalent cationic lipid (MLV5), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and DSPE-PEG, termed multi-LP delivered poorly hydrophilic α-GalCer adjuvants. Systemic administration and vaccination of multi-LP/α-GalCer with TRP2-mRNA induced strong tumor growth inhibitory effects. Moreover, the potency of mRNA vaccines against poor immunogenic and highly aggressive B16-F10 murine melanoma models was modified significantly (Guevara et al. 2019).
Polypeptides and cell-penetrating peptides (CPPs)
Polycationic peptides condense mRNA through electrostatic interaction. This process increases enzymatic tolerance and facilitates target tumor cell entry. These peptides could lower the risk of unfavorable pharmacokinetics and immune-stimulatory properties of mRNA. Wang et al. (2012) conducted IVT-mRNA transfection through peptides The core Protamine-mRNA conjugates were surrounded by PEGylated lipid and functionalized with a low molecular weight anisamide (AA). Based on the size and transfection efficiency, the optimized LPR-6 prolonged the half-lives in a biological system. It showed a robust fluorescence signal that improved tumor accumulation without elevation of inflammatory markers. Highly efficient LPR-6 to transfect GFP in H460 cells showed similar trends in a mouse model. It induced expression of suicidal gene HSV1-tk/ganciclovir (GCV). Therefore, superior tumor retarding effects were seen in the H460 xenograft animal model (Wang et al. 2013). In another example of cell-penetrating peptides, an amphipathic RALA motif with the α-helical structure of arginine and leucine residues where cationic nitrogen of peptides reacts with anionic phosphates of mRNA mediates the expression of encoded mRNA inside the DCs cell. Modified OVA mRNA-RALA nanocomplexes instigate cytolytic T cell responses compared to unmodified mRNA complexes. Amphipathic motifs in CPPs showed a complete loss of their transfection efficiency. Further, outperformed RALA mediated modified mRNA immunization over the classical liposomal-mRNA conjugate reduced the type I IFN responses that relieve the blocking of cytotoxic T cell responses (Udhayakumar et al. 2017). However, Bell et al. (2018) introduced a new CPP variant, Xentry. This structure penetrates adherent cells and delivers macromolecules, including peptides siRNA fused with human protamine (XP). The action facilitates the transfection efficiency of mRNA in various cell lines. Subsequently,, an addition of TLRs (toll-like receptors) antagonist 6-[3-(pyrrolidin-1-yl) propoxy)-2-(4-(3-(pyrrolidin-1-yl) propoxy) phenyl] benzo[d] oxazole (E6446) with XP induced the expression of cystic fibrosis transmembrane regulator (CFTR) protein into epithelial cells with 5–20 fold reduction of TLR 7 and 9 activations than other antagonists chloroquine (CQ) and hydroxychloroquine (HCQ) treating groups (Bell et al. 2018).
Coolen et al. (2019) conducted a comparative study on three different CPPs analog RALA, LAH4, and LAH4-L1 on transfecting DCs and stimulating immune signaling responses. Among the variants, LAH4-L1, by replacing histidines and leucines, showed notable transfection of an encoded mRNA in DC2.4 cells. The structure activated innate and immune signaling responses. Poly (lactic acid) nanoparticles (PLANPs) vectoring LAH4-L1/mRNA fastened mRNA release after clathrin-mediated endocytosis inside the Dc cells. This action occurs through the protonation of the histidine imidazole groups in LAH4-L1. However, the system failed to transfect mRNA expression in HEK293 and HeLa cells due to insignificant endocytosis capacities in the two epithelial cells (Coolen et al. 2019). More recently, He et al. (2021) investigated the effect of structural and biophysical differences of peptides vector on mRNA stability and transfection efficiency. Histidine-lysine (HK) peptides, H3K4b and its analog H3K(+ H)4b, H3k(+ H)4b, H-H3K(+ H) 4b, HH-H3K(+ H)4b, and H4K4b were designed by inserting different histidine motif in the second domain of HK peptides. An H3K (+ H)4b had one additional histidine incorporated in the terminal branch’s second -HHHK- motif. The new structure (H3K4b) exerted superior stability and cellular internalization. In conjunction with liposome (H3K(+ H) 4b-mRNA polyplex), it exerted ten times more luciferase expression in MDA-MB-231 cells. The authors argued that extra histidine in peptide structure and conjugation with DOTAP liposome boosts the vectoring efficiency of mRNA towards the target cell by lowering enzymatic degradation (He et al. 2021).
Virus-like particles (VLPs)
These molecules are primarily incorporated into the conventional mRNA transporter to halt enzymatic degradation and transfection efficiency. For example, in the wide-ranged therapeutic application of cell-penetrating peptides, protamine in preclinical and clinical studies was prevented by poor delivery efficiency associated with enzymatic degradation and low cross-linking capacity. Bacteriophage PP7 virus-like particles (VLPs) were customized with low molecular weight protamine (LMWP) for effective intracellular delivery of mRNA. The recombinant 2PP7-Protamine-GFP VLP can easily be translated into mature protein in mammalian cells by inserting the GFP gene between the XbaI and BamHI restriction sites (Sun et al. 2016). Jekhmane et al. (2017) constructed an artificial viral coat protein having well-tolerated transfection efficiency of pDNA and inspected their ability to stabilize and translational efficiency of mRNA. The artificial triblock viral coat protein, C-S10-K12, comprised C-terminal oligolysine (K12) for mRNA condensation. Silk-like midblock S10 is the backbone that self-assembles with mRNA to form rod shape viral-like particles and a long hydrophilic random coil polypeptide with a high proportion of glycines and prolines for colloidal stability. VLPs encapsulate the mRNA with adequate protection against enzymatic degradation and yield cellular expression of green fluorescent protein (EGFP) and luciferase in HEK293 cells, although not more than conventional liposome formulation. The data showed disparities that could be improved by incorporating rational cellular uptake and quick endosomal escape mediating agents (Jekhmane et al. 2017). Another study examined VSVG-L7Ae VLP synthesis combining envelope protein G of Vesicular stomatitis virus (VSV-G) and RNA binding protein. L7Ae of Archeoglobus fulgidus transported mRNA in hard to transfect cell lines and resulted in a significant expression of EGFP in human induced pluripotent stem cells (iPS cells) and monocytes. Furthermore, the insertion of mRNA into VSVG-L7Ae significantly increased through kink-turn motifs. This action triggered adequate internalization and transgene expression of encoded mRNA compared to bacteriophage MS2-binding protein (MS2BP) VSV-G (Zhitnyuk et al. 2018).
Clinical trials with mRNAs
Production of desired therapeutic protein in the human body by IVT mRNA drugs is more advantageous than the traditional fermentation process of protein manufacturing. Over the last two decades, several clinical investigations attempted to deliver therapeutic mRNA for cancer immunotherapy with ex-vivo immune cell transfection and re-infusion. This technique continues to be tested in more than 20 clinical trials. Most of these trials use the traditional approach of transferring dendritic cells electroporated with antigens relevant to tumor development. However, recent clinical trials focus on genomic sequencing to unveil tumor antigens specific to patients to develop personalized care immunotherapies for brain tumors. Further, T cells are also transfected with tumor-targeting CARs to treat cancers, including mesothelioma, B cell lymphoma, and B cell leukemia (Hajj and Whitehead 2017).
Besides immunotherapy, numerous mRNA-based protein therapies demonstrated significant clinical implications for secreted and intracellular proteins (Table 1). In phase I trials, Moderna developed mRNA targeting the OX40-binding partner (OX40L), a tumor necrosis factor receptor (TNFR) family member, and TNF superfamily that mediates the activation of CD4 and CD8 T cells, in addition to other lymphocytes and non-lymphocytes (Kowalski et al. 2019). During phase II clinical trials, Moderna and its partner AstraZeneca examined VEGF mRNA delivery to promote heart regeneration following myocardial infarction. Meanwhile, Translate Bio investigated mRNA encoding CFTR delivery (cystic fibrosis transmembrane conductance regulator) to the lungs via lipid nanoparticles in Phase I/II clinical studies of cystic fibrosis. Conry et al. (1995) generated the first mRNA cancer vaccine (Conry et al. 1995). Recently, German companies, Ethos151 and BioNTech152, are conducting clinical trials of mRNA-based therapies for cystic fibrosis and tumor immunotherapy (Kowalski et al. 2019). The process involves injecting mRNA coding for carcinoembryonic antigen (CEA) into mice muscles. The naked mRNA triggers antitumor adaptive immune responses. Since mRNA degrades quickly, clinical efficacy is minimal. Clinical trials utilize mRNA in nanosystems delivery for in vivo application. The most common are lipids. In an attempt to quickly respond to infectious disease outbreaks, lipid nanoparticle–mRNA formulations have emerged as an efficient and effective vaccine platform (Gómez-Aguado et al. 2020; Gebre et al. 2021; Kim et al. 2021). However, the unstable nature and short half-life of mRNA warrant detailed examination.
Before the clinical use of lipid nanoparticles, it is imperative to determine safety issues and storage conditions. Several cancer vaccines based on lipid nanoparticle–mRNA compositions are currently undergoing clinical trials (Table 1). For instance, FixVac encodes four non-mutated antigens of melanoma, developed with the RNA-LPX formulation. Based on the interim results from the phase I trial (NCT02410733), after the sixth immunization, the significant improvement in metabolism in the spleen provides evidence of FixVac delivery and immune cell activation. Eight immunizations have elicited immunity in approximately 75% of patients against one or more antigens linked to cancer, with CD8+ T cells as a vital player in T cell responses (Sahin et al. 2020). FixVac’s anticancer activity increases with an anti-programmed cell death protein 1 (PD1) antibody, leading to a 35% tumor growth inhibition rate in patients treated with immune checkpoint inhibitors (Sahin et al. 2020). In another study, mRNA-4157 encoded 34 neoantigens (NCT03897881) among patients with resected or unresectable solid tumors. The results of a phase I study (NCT03897881) were analyzed to determine the immunogenic properties of mRNA-4157 alone or in conjunction with immune checkpoint inhibitors. Following an average follow-up duration of 8 months, 14 out of 16 patients treated with monotherapy remained disease-free throughout the study. Overall, 50% of the combination groups (negative for human papillomavirus, non-immune checkpoint inhibitor, head and neck squamous cell cancer) responded to the therapy and exhibited a 9.8-month median progression-free survival (Burris lii 2019).
mRNA immunizations using non-viral vectors are currently being studied, even though dendritic cells (DC)-based therapeutics still represent most mRNA immunizations. T-cell activation was observed in patients with gastrointestinal cancer following mRNA immunization with a similar lipid nanoparticle composition (NCT03480152) (Cafri et al. 2020). CureVac, BioNTech, and Moderna announced the development of many mRNA formulations with several ongoing preclinical and clinical trials (Crommelin et al. 2021; Kowalski et al. 2019). Other prominent pharmaceutical companies, including Genentech, Amgen, and Merck, have conducted more mRNA vaccine trials (Mullard 2016). Moderna’s cytomegalovirus (CMV) vaccine, mRNA-1647, includes six mRNAs that encode various viral proteins. Five proteins comprise the CMV gH Pentamer complex and the herpesvirus glycoprotein (Table 1). BioNTech and Moderna have recently studied the delivery of mRNA against the Zika virus (Pardi et al. 2017; Richner et al. 2017). Low-dose vaccinations can prevent infections caused by viruses. The two mRNA vaccines developed for COVID-19, mRNA-1273 (NCT04470427) (Anderson et al. 2020; Baden et al. 2020) and BNT162b2 (NCT04368728) (Polack et al. 2020), have been rapidly developed and clinically assessed. Both vaccines use ionizable lipid nanoparticles to deliver full-length spike proteins encoded by SARS-CoV-2. In clinical trials, specific lipid nanoparticle–mRNA vaccine formulations are being investigated as influenza vaccines (Brazzoli et al. 2016; Bahl et al. 2017; Feldman et al. 2019; Perche et al. 2019) (Table 1). Influenza viruses require haemagglutinin as a surface antigen. Previous researchers have completed phase I clinical trials with mRNA-1440 and mRNA-1851, incorporating lipid nanoparticles and mRNA encoding haemagglutinin from the H10N8 and H7N9 influenza viruses, respectively (NCT03076385 and NCT03345043 (Bahl et al. 2017; Feldman et al. 2019). Currently, an antibody against Chikungunya virus (CHKV-IgG) is being produced in vivo using mRNA-1944, a lipid nanoparticle formulation containing mRNA (NCT03829384) (Hou et al. 2021). Moderna also intends to conduct two cancer immunotherapy trials, one of which will be in conjunction with Merck. In collaboration with Moderna, AstraZeneca has submitted a clinical trial application to begin phase I trials employing VEGFA-encoding mRNA formulations to stimulate angiogenesis in heart patients (Ding et al. 2014).
Table 1 shows the clinical trials for mRNA therapies analyzed in previous studies.
Translational challenges of mRNA
IVT mRNA systemic delivery challenges
Intravenously administered mRNA drugs in systemic circulation face multiple hurdles before reaching target cells. The backbone of mRNA is susceptible to quick degradation by nuclease enzymes in biological fluids, skin, and blood. On the other hand, NPs masking enzymatic degradation in the systemic circulation has the potential to access liver cells, spleen cells, bone marrow, and RES through fenestrated or non-fenestrated capillaries due to their small particle size (Wang et al. 2012). Moreover, the NPs-mRNA complex in blood encounters opsonin proteins from the physiological environment, namely, blood, interstitial fluid, and cellular cytoplasm. A new synthetic NP-PC (nanoparticles-protein corona) complex is formed and alters the pharmacokinetics and bio-distribution pattern of the NPs inside the body. The uneven bio-distribution results in unwanted side effects.. An additional dose of drugs is required to get optimum therapeutic action that increases overall treatment cost. In addition, specific delivery of NP-mRNAs into the brain, myocardial cells and cancer cells is also facing challenges as they have limited accessibility to cross BBB and abnormal tumor microenvironments. After the internalization of NP-mRNA complex into target cells, the release of mRNA from delivery cargo at the endosomal stage is another issue that must be addressed (Fig. 3). A study revealed a lower endosomal escape rate of < 0.01% every 15 min, even at higher cellular uptake of siRNA, more than 100,000 every two hours (Dowdy 2017; Juliano 2016; Wittrup et al. 2015). In this regard, several research groups have proposed multiple strategies to fasten the release drugs or genetic materials from delivery platforms for quick pharmacological responses to prevent drug loss inside the cell. Chowdhury (2013) developed inorganic pH-responsive carbonate apatite nanoparticles protonated with early endosome acidic pH This process releases the drug or RNAi therapeutics by disrupting endosomal membrane. Vaidyanathan et al. (2016a, b) reported a detailed mechanism of cellular uptake pathways of polymeric nanoparticle-based DNA into cells and their release pattern from the endosome. Cationic polymeric nanoparticles endocytose into the cell by lipid recycling pathways. Then, the intercalation of the endosomal membrane releases the payloads into the cytosol. This process increases the permeability of the endosomal membrane.

Primary translational challenges of IVT mRNA
GMP compliance
Up-scaling industrial manufacturing is still challenging, despite various advantages of mRNA therapeutics, such as successful rapid response manufacture and clinical approval of COVID-19. Maintaining GMP at the laboratory scale is mandatory to bridge the bench-bed gap. The rapid manufacture of IVT mRNA from a plasmid DNA vector involves an open reading frame promoter (ORF), UTRs, and poly-A Tail with the help of recombinant enzymes and ribonucleotide triphosphates (NTPs). However, the rarity of good laboratory practices (GLPs), quality of preclinical results, and reproducibility hinder regulatory approval and commercialization of optimized mRNA therapeutics. Every synthetic process, from DNA template production to purification, should be performed by adopting GLPs at laboratory scales for the GMP production of mRNA. The mRNA synthetic kits, e.g., plasmid DNA, Enzyme, and NTPs purchased from authorized and certified vendors, must be graded clinically for a translatable construct.
Based on Fig. 3, the six points must be scrutinized before clinical trials. Proper investigation on the selection and design of mRNA delivery vehicles lowers the rejection rate in clinical settings.
The final IVT mRNA drug constructs must pass a series of GMP standard tests, i.e., characterization, mRNA encapsulation efficiency, potency, sterility, and stability. Finally, the formulated mRNA drugs must be transferred into a sterile vial with an RNases-free storage buffer for prolonged use. Overall, the implication of productive mRNA synthesis, academic-industrial collaboration, and application of translational sciences propel the transformation of academically discovered therapeutics at a reasonable cost.
Scalability
Another obstacle to overcome for the commercialization of mRNA drugs is switching the scalable production of mRNA from small laboratory operations to mass commercial production. Unique physicochemical properties, drug content, integrity, and batch to batch reproducibility affect commercial mRNA drugs (Pascolo 2004; Geall et al. 2013). Further, the SOP for synthesizing mRNA and purification steps should be optimized by adopting GMPs from regulatory authorities. Scientific gaps, technology, physicochemical characterization, preclinical models, toxicity, and potential risk in human trials must be assessed. Hence, mRNA production is prolific, with approximately 2gl−1 of mRNA produced from multi-gram reactions (Pardi et al. 2018). The number, extent, rate and kinetics of mRNA translation and the number of targeted cells should be investigated based on the type and severity of respective diseases. At the time of writing, two nanoparticle-based mRNA vaccines from Pfizer and Moderna have been approved, and another 12 clinical trials of mRNA-based vaccines are ongoing. The clinical results will provide valuable huge human data on dose, efficacy, and toxicity (Verbeke et al. 2021). By capitalizing on the scientific data, the selection, design, and manufacture of mRNA-based therapeutics can be studied for multiple diseases and cancers. Massive data on pharmaceutical technology, manufacturing, quality control, stability and storage of NP-based mRNA vaccine increases mRNA mass production through different drug delivery routes. The established delivery routes are Intravenous, intraperitoneal, Intranasal, subcutaneous, and systemic routes. Different dosages of mRNA vaccines are applied through these routes to different organs, such as the brain, liver, and lungs.
Regulatory features
Since mRNA therapeutics is still in the infancy stage of clinical trials, there are no specific regulatory guidelines from the governing body for preclinical and clinical studies, industrial manufacturing, and post-marketing surveillance studies except for COVID-19 vaccines approved on an emergency basis. The diversified therapeutic applications of mRNA biologics as prophylaxis in cancer treatment warrant more specific regulations from classification to pharmaceutical manufacturing. In this regard, the regulatory framework for commercialization focuses on production, quality control, technologies, and safety of mRNA therapeutics. More efforts should be taken to establish detailed guidelines by national and international agencies, such as WHO, US-FDA, and European medicine agencies to select industrial-grade materials, crucial manufacturing steps, purification methods, and potency evaluation. For preclinical and clinical studies, specific rules must be implemented to monitor systemic toxicity, potential adverse inflammatory adverse effects, dose, and tolerability. A comprehensive regulatory guideline should also be established to quantify the mRNA encapsulation rate, particle size, distribution of mRNA-NPs complex, and thermal stability to ensure batch-to-batch reproducibility. Therefore, the consensus from international and strategic advisory authorities will provide a roadmap to develop and evaluate mRNA-based therapeutics for treating cancers and other diseases in the future.
Safety concern
As the prospects of mRNA are appealing in modern prophylactic vaccines, safety profile, and tolerability must be strictly maintained. The rapid and cell-free mRNA manufacturing steps and its non-integrative nature in cells provide additional advantages over conventional pDNA vaccines and recombinant proteins. However, systemic administration of mRNA induces inflammatory responses by triggering TLR3, TLR7, TLR8, Interferon-α, IL-6, and cytokines (Nestle et al. 2005; Theofilopoulos et al. 2005). Moreover, viral and non-viral mRNA delivery vectors trigger systemic inflammation and activate autoimmune antibodies. These structures also have poor and non-specific biodistribution profile and transporter components linked to toxicity. Extracellular mRNA causes edema, blood coagulation, and pathological thrombus formation by increasing the permeability of endothelial cells (Kannemeier et al. 2007; Redzic et al. 2014). Although there is massive global demand for the mRNA vaccination program, evaluating the safety and immunogenicity profiles of mRNA prophylactic and therapeutic applications is advisable. Nevertheless, more clinical data and documented guidelines to assess the safety and toxicity of mRNA drugs will be available for the future development of optimal mRNA drugs for different treatment purposes.
Shelf-life and storage conditions
The shelf-life and storage conditions, such as temperature, storage buffer, or reagents of finished mRNA drugs, are the most critical and unresolved challenges facing mRNA manufacturers. At ambient temperature, the efficiency of lipid-based mRNA vaccines declined. Therefore, an optimum temperature of -15℃ to -80℃ preserves lipid-based mRNA vaccines within their shelf-life during transportation. For example, mRNA-1273 from Moderna must be stored at − 15 to − 20 °C and BNT162b2 from BioNTech/ Pfizer at − 60 °C to − 80 °C for storage (Zhang et al. 2022a, b, c). The cold storage temperature of mRNA drugs creates severe logistic problems in stockpiling and distribution, particularly in countries lacking cold-chain infrastructure. In order to resolve the issue, first, an ideal nanoparticle/mRNA complexes stable at ambient temperature should be designed, followed by a formulation buffer or cryoprotectant that may increase the stability of mRNA drugs for long-term storage.
Future recommendations
Since the first in-vivo mRNA delivery in 1990, mRNA-based therapeutics have gained significant attention in biopharmaceuticals as a next-generation genetic medicine. The breakthrough in NPs-based mRNA vaccines against COVID-19 has increased the development of mRNA therapies for fighting cancer and other genetic diseases not treated with current treatment modalities. mRNA effectively conveys the coded message to the target cell. It induces transient protein expression without the risk of insertional mutagenesis. The success and versatility of mRNA therapeutics ranging from cancer to COVID-19 are hindered by overriding factors, such as biological instability, cellular trafficking, and activation of TLR3/7 triggered innate immune system. However, considerable progress has been made in modifying the structural features of IVT mRNA. Clinically advanced nanomaterials may improve pharmacological actions for selective delivery to the target cytoplasm. The significant number of ongoing preclinical and clinical studies for a wide range of incurable and genetic diseases at the laboratory and industrial level indicates a growing interest among the pharmaceutical industry, sponsors, and policymakers in mRNA drugs. The rapid development of NPs-mRNA based COVID-19 vaccine and ongoing clinical trial at phase 4 may answer unresolved issues, such as “unknown risks of nanomedicine and how nanoparticles behave inside the body.” However, better efforts in each of the following areas should be made to improve mRNA drug tolerance, efficacy, and delivery.
mRNA design
The first initiative to select ideal mRNA constructs for cancer management is to understand the molecular mechanism of cancer development and identify key cancer-causing genes responsible for cancer development and metastasis. After discovering the respective genes, mRNA could be designed to increase the gene expression or induce immune responses against the tumor. Secondly, by studying the SAR (Structure–activity relationship) of IVT mRNA, serial optimization and modification within CAP structures, UTRs, poly-A tail, and addition of modified bases should be considered to develop stable, tolerable next-generation mRNA drugs with maximum translational efficiency. To get quick clinical approval, the content of impurities during IVT production, standard quality control, and industrial flexible purification methods may need to be assured. Moreover, scientists may focus on repurposing the same fundamental building block of mRNA to treat different diseases simply by changing the coding sequence. Finally, more attempts should be required to develop the synthetic economic method, improvised manufacturing technology to ensure high yield pharmaceutical production of mRNA at a reasonable cost.
Selection of delivery materials
Based on the current scenario, many transporters have been tested in preclinical and clinical studies to deliver mRNA for cancer treatment. It is not practical to administer naked mRNA into the biological system. However, the number and rate of clinical approvals are few compared to animal studies. Therefore, scientists should focus on the following aspects:
-
(1)
Nanocarriers synthesized from FDA-approved materials to propel rapid succession;
-
(2)
The Substantial improvements in viable industrial manufacturing, formulation steps, and biodegradability of nanocarriers;
-
(3)
Adequate stability of NPs-mRNA complexes in systemic circulation and the efficient targeting capacity to avoid off-target distribution and drug loss;
-
(4)
The mechanistic pathways of cellular internalization and release mechanism from delivery cargoes at endosomal stages should be clarified as it is fundamental for the pharmacological action of drugs;
-
(5)
The dose and toxicity of nanocarriers should be optimized by analyzing data from animal and clinical trials.
By acquiring adequate human data from ongoing clinical studies of NPs-based mRNA vaccines and translational science to bridge the preclinical and clinical gap, the scientific community can introduce next-generation NPs-mRNA drugs to eliminate cancer.
Conclusion
mRNA has a unique and transient expression that lowers the risk of insertional mutagenesis. Since the discovery of in vitro transcribed mRNA, synthetic mRNA has gained tremendous interest from different biology, medicine, and material science scientists. Diverse applications of mRNA therapeutics have revolutionized gene editing, protein replacement therapies, cell reprogramming, and immunotherapies. Further, substantial progress in mRNA design, structure, and chemical modifications overcomes the instability and immunogenicity of mRNA drugs. Although the whole world has witnessed a new era of mRNA vaccines to fight against the unprecedented COVID-19 pandemic, some key challenges must be addressed for the expansion of mRNA technology to treat many genetic diseases like cancer. The primary challenge includes discovering reasonable and scalable mRNA synthetic procedures compatible with existing pharmaceutical technologies. Potent mRNA therapeutics depends on clinically translatable mRNA transporters, adequate stability in the systemic circulation, and resolution of immunogenicity. In conclusion, clinically advanced nanomaterials improve mRNA-based nanotherapeutics for cancer and neurodegenerative treatment.
Data availability
The authors declare that the main data are available within the article.
References
Anderson DG, Lynn DM, Langer R (2003) Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angewandte Chemie Intern Ed Engl 42(27):3153–3158. https://doi.org/10.1002/anie.200351244
Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D, Karikó K (2010) Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucl Acids Res 38(17):5884–5892. https://doi.org/10.1093/nar/gkq347
Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Karikó K (2011) Nucleoside modifications in RNA limit activation of 2’-5’-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucl Acids Res 39(21):9329–9338. https://doi.org/10.1093/nar/gkr586
Anderson EJ, Rouphael NG, Widge AT, Jackson LA, Roberts PC, Makhene M, Chappell JD, Denison MR, Stevens LJ, Pruijssers AJ, McDermott AB, Flach B, Lin BC, Doria-Rose NA, O’Dell S, Schmidt SD, Corbett KS, Swanson PA, Padilla M, Neuzil KM, Bennett H, Leav B, Makowski M, Albert J, Cross K, Edara VV, Floyd K, Suthar MS, Martinez DR, Baric R, Buchanan W, Luke CJ, Phadke VK, Rostad CA, Ledgerwood JE, Graham BS, Beigel JH (2020) Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N Engl J Med 383(25):2427–2438. https://doi.org/10.1056/NEJMoa2028436
Andries O, De Filette M, De Smedt SC, Demeester J, Van Poucke M, Peelman L, Sanders NN (2013) Innate immune response and programmed cell death following carrier-mediated delivery of unmodified mRNA to respiratory cells. J Control Release 167(2):157–166. https://doi.org/10.1016/j.jconrel.2013.01.033
Arya S, Lin Q, Zhou N, Gao X, Huang JD (2020) Strong immune responses induced by direct local injections of modified mRNA-lipid nanocomplexes. Mole Ther Nucl Acids 19:1098–1109. https://doi.org/10.1016/j.omtn.2019.12.044
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T (2020) Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med 384(5):403–416. https://doi.org/10.1056/NEJMoa2035389
Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, Laska ME, Smith M, Almarsson Ö, Thompson J, Ribeiro AM, Watson M, Zaks T, Ciaramella G (2017) Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol Ther 25(6):1316–1327. https://doi.org/10.1016/j.ymthe.2017.03.035
Baptista B, Carapito R, Laroui N, Pichon C, Sousa F (2021) mRNA, a revolution in biomedicine. Pharmaceutics. https://doi.org/10.3390/pharmaceutics13122090
Barbier AJ, Jiang AY, Zhang P, Wooster R, Anderson DG (2022) The clinical progress of mRNA vaccines and immunotherapies. Nat Biotechnol 40(6):840–854. https://doi.org/10.1038/s41587-022-01294-2
Bell GD, Yang Y, Leung E, Krissansen GW (2018) mRNA transfection by a Xentry-protamine cell-penetrating peptide is enhanced by TLR antagonist E6446. PLoS ONE 13(7):e0201464. https://doi.org/10.1371/journal.pone.0201464
Bhavsar MD, Amiji MM (2008) Development of novel biodegradable polymeric nanoparticles-in-microsphere formulation for local plasmid DNA delivery in the gastrointestinal tract. AAPS PharmSciTech 9(1):288–294. https://doi.org/10.1208/s12249-007-9021-9
Brazzoli M, Magini D, Bonci A, Buccato S, Giovani C, Kratzer R, Zurli V, Mangiavacchi S, Casini D, Brito LM, De Gregorio E, Mason PW, Ulmer JB, Geall AJ, Bertholet S (2016) Induction of broad-based immunity and protective efficacy by self-amplifying mRNA vaccines encoding influenza virus hemagglutinin. J Virol 90(1):332–344. https://doi.org/10.1128/jvi.01786-15
Brenner S, Jacob F, Meselson M (1961) An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 190:576–581. https://doi.org/10.1038/190576a0
Brito LA, Kommareddy S, Maione D, Uematsu Y, Giovani C, Berlanda Scorza F, Otten GR, Yu D, Mandl CW, Mason PW, Dormitzer PR, Ulmer JB, Geall AJ (2015) Self-amplifying mRNA vaccines. Adv Genet 89:179–233. https://doi.org/10.1016/bs.adgen.2014.10.005
Burris Iii HA, Patel MR, Cho DC, Clarke JM, Gutierrez M, Zaks TZ, Frederick J, Hopson K, Mody K, Binanti-Berube A, Robert-Tissot C, Cowens K, Breton B, Zhong S, Zhou H, Cohen PS, Keating K, Meehan RS, Gainor JF (2019) A phase 1, open-label, multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in subjects with resected solid tumors and in combination with pembrolizumab in subjects with unresectable solid tumors (Keynote-603). J Glob Oncol 5(suppl):93. https://doi.org/10.1200/JGO.2019.5.suppl.93
Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, Parkhurst MR, Yossef R, Lowery FJ, Jafferji MS, Prickett TD, Goff SL, McGowan CT, Seitter S, Shindorf ML, Parikh A, Chatani PD, Robbins PF, Rosenberg SA (2020) mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Investig 130(11):5976–5988. https://doi.org/10.1172/jci134915
Calero G, Wilson KF, Ly T, Rios-Steiner JL, Clardy JC, Cerione RA (2002) Structural basis of m7G pppG binding to the nuclear cap-binding protein complex. Nat Struct Mol Biol 9(12):912–917. https://doi.org/10.1038/nsb874
Calvo O, Manley JL (2003) Strange bedfellows: polyadenylation factors at the promoter. Genes Dev 17(11):1321–1327. https://doi.org/10.1101/gad.1093603
Capasso Palmiero U, Kaczmarek JC, Fenton OS, Anderson DG (2018) Poly(β-amino ester)-co-poly(caprolactone) terpolymers as nonviral vectors for mRNA delivery in vitro and in vivo. Adv Healthcare Mater 7(14):e1800249. https://doi.org/10.1002/adhm.201800249
Chabeda A, Yanez RJR, Lamprecht R, Meyers AE, Rybicki EP, Hitzeroth II (2018) Therapeutic vaccines for high-risk HPV-associated diseases. Papillomavirus Res 5:46–58. https://doi.org/10.1016/j.pvr.2017.12.006
Chen Q, Qi R, Chen X, Yang X, Wu S, Xiao H, Dong W (2017) A targeted and stable polymeric nanoformulation enhances systemic delivery of mRNA to tumors. Mol Ther 25(1):92–101. https://doi.org/10.1016/j.ymthe.2016.10.006
Cheng C, Convertine AJ, Stayton PS, Bryers JD (2012) Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials 33(28):6868–6876. https://doi.org/10.1016/j.biomaterials.2012.06.020
Cherng JY, van de Wetering P, Talsma H, Crommelin DJ, Hennink WE (1996) Effect of size and serum proteins on transfection efficiency of poly ((2–dimethylamino)ethyl methacrylate)–plasmid nanoparticles. Pharm Res 13(7):1038–1042. https://doi.org/10.1023/a:1016054623543
Chowdhury EH (2013) pH-responsive magnesium- and carbonate-substituted apatite nano-crystals for efficient and cell-targeted delivery of transgenes. Open J Genet 3(2A):33687. https://doi.org/10.4236/ojgen.2013.32A1005
Conry, R. M., LoBuglio, A. F., Wright, M., Sumerel, L., Pike, M. J., Johanning, F., Benjamin, R., Lu, D., Curiel, D. T. (1995). Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Research, 55(7), 1397–1400. http://www.scopus.com/inward/record.url?scp=0028965192&partnerID=8YFLogxK
Coolen A-L, Lacroix C, Mercier-Gouy P, Delaune E, Monge C, Exposito J-Y, Verrier B (2019) Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 195:23–37. https://doi.org/10.1016/j.biomaterials.2018.12.019
Crommelin DJA, Anchordoquy TJ, Volkin DB, Jiskoot W, Mastrobattista E (2021) Addressing the cold reality of mRNA vaccine stability. J Pharm Sci 110(3):997–1001. https://doi.org/10.1016/j.xphs.2020.12.006
Cu Y, Broderick KE, Banerjee K, Hickman J, Otten G, Barnett S, Kichaev G, Sardesai NY, Ulmer JB, Geall A (2013) Enhanced delivery and potency of self-amplifying mRNA vaccines by electroporation in situ. Vaccines. https://doi.org/10.3390/vaccines1030367
de Maeyer-Guignard J, de Maeyer E, Montagnier L (1972) Interferon messenger RNA: translation in heterologous cells. Proc Natl Acad Sci USA 69(5):1203–1207. https://doi.org/10.1073/pnas.69.5.1203
Debus H, Baumhof P, Probst J, Kissel T (2010) Delivery of messenger RNA using poly(ethylene imine)-poly(ethylene glycol)-copolymer blends for polyplex formation: biophysical characterization and in vitro transfection properties. J Control Release 148(3):334–343. https://doi.org/10.1016/j.jconrel.2010.09.007
Dhaliwal HK, Fan Y, Kim J, Amiji MM (2020) Intranasal delivery and transfection of mRNA therapeutics in the brain using cationic liposomes. Mol Pharm 17(6):1996–2005. https://doi.org/10.1021/acs.molpharmaceut.0c00170
Dimitriadis GJ (1978) Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 274:923–924. https://doi.org/10.1038/274923a0
Ding Y, Jiang Z, Saha K, Kim CS, Kim ST, Landis RF, Rotello VM (2014) Gold nanoparticles for nucleic acid delivery. Mol Ther 22(6):1075–1083. https://doi.org/10.1038/mt.2014.30
Dong Y, Love KT, Dorkin JR, Sirirungruang S, Zhang Y, Chen D, Bogorad RL, Yin H, Chen Y, Vegas AJ, Alabi CA, Sahay G, Olejnik KT, Wang W, Schroeder A, Lytton-Jean AK, Siegwart DJ, Akinc A, Barnes C, Barros SA, Carioto M, Fitzgerald K, Hettinger J, Kumar V, Novobrantseva TI, Qin J, Querbes W, Koteliansky V, Langer A, D. G. (2014) Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc Natl Acad Sci USA 111(11):3955–3960. https://doi.org/10.1073/pnas.1322937111
Dong Y, Dorkin JR, Wang W, Chang PH, Webber MJ, Tang BC, Yang J, Abutbul-Ionita I, Danino D, DeRosa F, Heartlein M, Langer R, Anderson DG (2016) Poly(glycoamidoamine) brushes formulated nanomaterials for systemic siRNA and mRNA delivery in vivo. Nano Lett 16(2):842–848. https://doi.org/10.1021/acs.nanolett.5b02428
Dowdy SF (2017) Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol 35(3):222–229. https://doi.org/10.1038/nbt.3802
Feldman RA, Fuhr R, Smolenov I, Mick Ribeiro A, Panther L, Watson M, Senn JJ, Smith M, Almarsson Ӧ, Pujar HS, Laska ME, Thompson J, Zaks T, Ciaramella G (2019) mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37(25):3326–3334. https://doi.org/10.1016/j.vaccine.2019.04.074
Fleeton MN, Chen M, Berglund P, Rhodes G, Parker SE, Murphy M, Atkins GJ, Liljeström P (2001) Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J Infect Dis 183(9):1395–1398. https://doi.org/10.1086/319857
Furusawa M, Nishimura T, Yamaizumi M, Okada Y (1974) Injection of foreign substances into single cells by cell fusion. Nature 249(456):449–450. https://doi.org/10.1038/249449a0
Geall AJ, Mandl CW, Ulmer JB (2013) RNA: the new revolution in nucleic acid vaccines. Semin Immunol 25(2):152–159. https://doi.org/10.1016/j.smim.2013.05.001
Gebauer F, Hentze MW (2004) Molecular mechanisms of translational control. Nat Rev Mol Cell Biol 5(10):827–835. https://doi.org/10.1038/nrm1488
Gebre MS, Brito LA, Tostanoski LH, Edwards DK, Carfi A, Barouch DH (2021) Novel approaches for vaccine development. Cell 184(6):1589–1603. https://doi.org/10.1016/j.cell.2021.02.030
Gilboa E, Vieweg J (2004) Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev 199:251–263. https://doi.org/10.1111/j.0105-2896.2004.00139.x
Goldstrohm AC, Wickens M (2008) Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol 9(4):337–344. https://doi.org/10.1038/nrm2370
Gómez-Aguado I, Rodríguez-Castejón J, Vicente-Pascual M, Rodríguez-Gascón A, Solinís M, Del Pozo-Rodríguez A (2020) Nanomedicines to deliver mRNA: state of the art and future perspectives. Nanomaterials. https://doi.org/10.3390/nano10020364
Grudzien-Nogalska E, Stepinski J, Jemielity J, Zuberek J, Stolarski R, Rhoads RE, Darzynkiewicz E (2007) Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol 431:203–227. https://doi.org/10.1016/s0076-6879(07)31011-2
Guan S, Rosenecker J (2017) Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther 24(3):133–143. https://doi.org/10.1038/gt.2017.5
Guevara ML, Jilesen Z, Stojdl D, Persano S (2019) Codelivery of mRNA with α-galactosylceramide using a new lipopolyplex formulation induces a strong antitumor response upon intravenous administration. ACS Omega 4(8):13015–13026. https://doi.org/10.1021/acsomega.9b00489
Hajj KA, Whitehead KA (2017) Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat Rev Mater 2(10):17056. https://doi.org/10.1038/natrevmats.2017.56
He J, Xu S, Leng Q, Mixson AJ (2021) Location of a single histidine within peptide carriers increases mRNA delivery. J Gene Med 23(2):e3295. https://doi.org/10.1002/jgm.3295
Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 352(6292):1413–1416. https://doi.org/10.1126/science.aad9868
Hou X, Zaks T, Langer R, Dong Y (2021) Lipid nanoparticles for mRNA delivery. Nat Rev Mater 6(12):1078–1094. https://doi.org/10.1038/s41578-021-00358-0
Huotari J, Helenius A (2011) Endosome maturation. EMBO J 30(17):3481–3500. https://doi.org/10.1038/emboj.2011.286
Jahan S, Karim ME, Chowdhury EH (2021) Nanoparticles targeting receptors on breast cancer for efficient delivery of chemotherapeutics. Biomedicines. https://doi.org/10.3390/biomedicines9020114
Jarzębińska A, Pasewald T, Lambrecht J, Mykhaylyk O, Kümmerling L, Beck P, Hasenpusch G, Rudolph C, Plank C, Dohmen C (2016) A single methylene group in oligoalkylamine-based cationic polymers and lipids promotes enhanced mRNA delivery. Angewandte Chemie Intern Ed Engl 55(33):9591–9595. https://doi.org/10.1002/anie.201603648
Jekhmane S, de Haas R, da Silva P, Filho O, van Asbeck AH, Favretto ME, Hernandez Garcia A, Brock R, de Vries R (2017) Virus-like particles of mRNA with artificial minimal coat proteins: particle formation, stability, and transfection efficiency. Nucleic Acid Ther 27(3):159–167. https://doi.org/10.1089/nat.2016.0660
Jemielity J, Kowalska J, Rydzik AM, Darzynkiewicz E (2010) Synthetic mRNA cap analogs with a modified triphosphate bridge – synthesis, applications and prospects. New J Chem 34(5):829–844. https://doi.org/10.1039/C0NJ00041H
Jogani VV, Shah PJ, Mishra P, Mishra AK, Misra AR (2007) Nose-to-brain delivery of tacrine. J Pharm Pharmacol 59(9):1199–1205. https://doi.org/10.1211/jpp.59.9.0003
Johansson DX, Ljungberg K, Kakoulidou M, Liljeström P (2012) Intradermal electroporation of naked replicon RNA elicits strong immune responses. PLoS ONE 7(1):e29732. https://doi.org/10.1371/journal.pone.0029732
Juliano RL (2016) The delivery of therapeutic oligonucleotides. Nucleic Acids Res 44(14):6518–6548. https://doi.org/10.1093/nar/gkw236
Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl M-L, Sedding D, Massberg S, Günther A, Engelmann B, Preissner Klaus T (2007) Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA 104(15):6388–6393. https://doi.org/10.1073/pnas.0608647104
Karikó K, Ni H, Capodici J, Lamphier M, Weissman D (2004) mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem 279(13):12542–12550. https://doi.org/10.1074/jbc.M310175200
Karikó K, Buckstein M, Ni H, Weissman D (2005) Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23(2):165–175. https://doi.org/10.1016/j.immuni.2005.06.008
Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D (2008) Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16(11):1833–1840. https://doi.org/10.1038/mt.2008.200
Karim ME, Rosli R, Chowdhury EH (2017) Systemic delivery of nanoformulations of anti-cancer drugs with therapeutic potency in animal models of cancer. Curr Cancer Ther Rev 12(3):204–220. https://doi.org/10.2174/1573394712666160919101827
Karim ME, Tha KK, Othman I, Borhan Uddin M, Chowdhury EH (2018) Therapeutic potency of nanoformulations of siRNAs and shRNAs in animal models of cancers. Pharmaceutics. https://doi.org/10.3390/pharmaceutics10020065
Karim ME, Shetty J, Islam RA, Kaiser A, Bakhtiar A, Chowdhury EH (2019) Strontium sulfite: a new pH-responsive inorganic nanocarrier to deliver therapeutic siRNAs to cancer cells. Pharmaceutics. https://doi.org/10.3390/pharmaceutics11020089
Kauffman KJ, Webber MJ, Anderson DG (2016) Materials for non-viral intracellular delivery of messenger RNA therapeutics. J Control Release 240:227–234. https://doi.org/10.1016/j.jconrel.2015.12.032
Kim J, Eygeris Y, Gupta M, Sahay G (2021) Self-assembled mRNA vaccines. Adv Drug Deliv Rev 170:83–112. https://doi.org/10.1016/j.addr.2020.12.014
Kofler RM, Aberle JH, Aberle SW, Allison SL, Heinz FX, Mandl CW (2004) Mimicking live flavivirus immunization with a noninfectious RNA vaccine. Proc Natl Acad Sci USA 101(7):1951–1956. https://doi.org/10.1073/pnas.0307145101
Kowalska J, Lewdorowicz M, Zuberek J, Grudzien-Nogalska E, Bojarska E, Stepinski J, Rhoads RE, Darzynkiewicz E, Davis RE, Jemielity J (2008) Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 14(6):1119–1131. https://doi.org/10.1261/rna.990208
Kowalska J, Wypijewska del Nogal A, Darzynkiewicz ZM, Buck J, Nicola C, Kuhn AN, Lukaszewicz M, Zuberek J, Strenkowska M, Ziemniak M, Maciejczyk M, Bojarska E, Rhoads RE, Darzynkiewicz E, Sahin U, Jemielity J (2014) Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res 42(16):10245–10264. https://doi.org/10.1093/nar/gku757
Kowalski PS, Rudra A, Miao L, Anderson DG (2019) Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther 27(4):710–728. https://doi.org/10.1016/j.ymthe.2019.02.012
Kuhn AN, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Türeci Ö, Sahin U (2010) Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther 17(8):961–971. https://doi.org/10.1038/gt.2010.52
Kwon H, Kim M, Seo Y, Moon YS, Lee HJ, Lee K, Lee H (2018) Emergence of synthetic mRNA: in vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 156:172–193. https://doi.org/10.1016/j.biomaterials.2017.11.034
Lane CD, Marbaix G, Gurdon JB (1971) Rabbit haemoglobin synthesis in frog cells: the translation of reticulocyte 9 s RNA in frog oocytes. J Mol Biol 61(1):73–91. https://doi.org/10.1016/0022-2836(71)90207-5
Leppek K, Das R, Barna M (2018) Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19(3):158–174. https://doi.org/10.1038/nrm.2017.103
Lewis JD, Gunderson SI, Mattaj IW (1995) The influence of 5’ and 3’ end structures on pre-mRNA metabolism. J Cell Sci Suppl 19:13–19. https://doi.org/10.1242/jcs.1995.supplement_19.2
Li Y, Kiledjian M (2010) Regulation of mRNA decapping. Wiley Interdiscip Rev RNA 1(2):253–265. https://doi.org/10.1002/wrna.15
Li W, Szoka FC Jr (2007) Lipid-based nanoparticles for nucleic acid delivery. Pharm Res 24(3):438–449. https://doi.org/10.1007/s11095-006-9180-5
Li B, Luo X, Deng B, Wang J, McComb DW, Shi Y, Gaensler KM, Tan X, Dunn AL, Kerlin BA, Dong Y (2015) An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett 15(12):8099–8107. https://doi.org/10.1021/acs.nanolett.5b03528
Li B, Luo X, Deng B, Giancola JB, McComb DW, Schmittgen TD, Dong Y (2016) Effects of local structural transformation of lipid-like compounds on delivery of messenger RNA. Sci Rep 6:22137. https://doi.org/10.1038/srep22137
Lorentzen CL, Haanen JB, Met Ö, Svane IM (2022) Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol 23(10):e450–e458. https://doi.org/10.1016/s1470-2045(22)00372-2
Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, Qin J, Cantley W, Qin LL, Racie T, Frank-Kamenetsky M, Yip KN, Alvarez R, Sah DW, de Fougerolles A, Fitzgerald K, Koteliansky V, Akinc A, Langer R, Anderson DG (2010) Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA 107(5):1864–1869. https://doi.org/10.1073/pnas.0910603106
Loyter A, Zakai N, Kulka RG (1975) “Ultramicroinjection” of macromolecules or small particles into animal cells. A new technique based on virus-induced cell fusion. J Cell Biol 66(2):292–304. https://doi.org/10.1083/jcb.66.2.292
Mayr C (2017) Regulation by 3’-untranslated regions. Annu Rev Genet 51:171–194. https://doi.org/10.1146/annurev-genet-120116-024704
Mays LE, Ammon-Treiber S, Mothes B, Alkhaled M, Rottenberger J, Müller-Hermelink ES, Grimm M, Mezger M, Beer-Hammer S, von Stebut E, Rieber N, Nürnberg B, Schwab M, Handgretinger R, Idzko M, Hartl D, Kormann MS (2013) Modified Foxp3 mRNA protects against asthma through an IL-10-dependent mechanism. J Clin Investig 123(3):1216–1228. https://doi.org/10.1172/jci65351
McIvor RS (2011) Therapeutic delivery of mRNA: the medium is the message. Mol Ther 19(5):822–823. https://doi.org/10.1038/mt.2011.67
McNamara MA, Nair SK, Holl EK (2015) RNA-based vaccines in cancer immunotherapy. J Immunol Res 2015:794528. https://doi.org/10.1155/2015/794528
Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH (2015) Therapeutic cancer vaccines. J Clin Investig 125(9):3401–3412. https://doi.org/10.1172/jci80009
Men K, Zhang R, Zhang X, Huang R, Zhu G, Tong R, Yang L, Wei Y, Duan X (2018) Delivery of modified mRNA encoding vesicular stomatitis virus matrix protein for colon cancer gene therapy. RSC Adv 8(22):12104–12115. https://doi.org/10.1039/C7RA13656K
Migliore MM, Vyas TK, Campbell RB, Amiji MM, Waszczak BL (2010) Brain delivery of proteins by the intranasal route of administration: a comparison of cationic liposomes versus aqueous solution formulations. J Pharm Sci 99(4):1745–1761. https://doi.org/10.1002/jps.21939
Migliore MM, Ortiz R, Dye S, Campbell RB, Amiji MM, Waszczak BL (2014) Neurotrophic and neuroprotective efficacy of intranasal GDNF in a rat model of Parkinson’s disease. Neuroscience 274:11–23. https://doi.org/10.1016/j.neuroscience.2014.05.019
Mignone F, Gissi C, Liuni S, Pesole G (2002) Untranslated regions of mRNAs. Genome Biol. https://doi.org/10.1186/gb-2002-3-3-reviews0004
Mockey M, Gonçalves C, Dupuy FP, Lemoine FM, Pichon C, Midoux P (2006) mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with poly(A) chains in cis and in trans for a high protein expression level. Biochem Biophys Res Commun 340(4):1062–1068. https://doi.org/10.1016/j.bbrc.2005.12.105
Mockey M, Bourseau E, Chandrashekhar V, Chaudhuri A, Lafosse S, Le Cam E, Quesniaux VF, Ryffel B, Pichon C, Midoux P (2007) mRNA-based cancer vaccine: prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Gene Ther Cancer 14(9):802–814. https://doi.org/10.1038/sj.cgt.7701072
Mukalel AJ, Riley RS, Zhang R, Mitchell MJ (2019) Nanoparticles for nucleic acid delivery: applications in cancer immunotherapy. Cancer Lett 458:102–112. https://doi.org/10.1016/j.canlet.2019.04.040
Mullard A (2016) The cancer vaccine resurgence. Nat Rev Drug Discov 15(10):663–665. https://doi.org/10.1038/nrd.2016.201
Nagy NA, de Haas AM, Geijtenbeek TBH, van Ree R, Tas SW, van Kooyk Y, de Jong EC (2021) Therapeutic liposomal vaccines for dendritic cell activation or tolerance. Front Immunol 12:674048. https://doi.org/10.3389/fimmu.2021.674048
Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu YJ, Gilliet M (2005) Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med 202(1):135–143. https://doi.org/10.1084/jem.20050500
Neugebauer KM (2002) On the importance of being co-transcriptional. J Cell Sci 115(Pt 20):3865–3871. https://doi.org/10.1242/jcs.00073
Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D (2017) Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett 17(3):1326–1335. https://doi.org/10.1021/acs.nanolett.6b03329
Painter MM, Mathew D, Goel RR, Apostolidis SA, Pattekar A, Kuthuru O, Baxter AE, Herati RS, Oldridge DA, Gouma S, Hicks P, Dysinger S, Lundgreen KA, Kuri-Cervantes L, Adamski S, Hicks A, Korte S, Giles JR, Weirick ME, McAllister CM, Dougherty J, Long S, D’Andrea K, Hamilton JT, Betts MR, Bates P, Hensley SE, Grifoni A, Weiskopf D, Sette A, Greenplate AR, Wherry EJ (2021) Rapid induction of antigen-specific CD4(+) T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity 54(9):2133-2142.e2133. https://doi.org/10.1016/j.immuni.2021.08.001
Palamà IE, Cortese B, D’Amone S, Gigli G (2015a) Gene therapy with nonviral poly(ε-caprolactone) nanoparticles. Ther Deliv 6(7):769–771. https://doi.org/10.4155/tde.15.41
Palamà IE, Cortese B, Damone S, Gigli G (2015b) mRNA delivery using non-viral PCL nanoparticles. Biomater Sci 3(1):144–151. https://doi.org/10.1039/C4BM00242C
Palucka AK, Coussens LM (2016) The basis of oncoimmunology. Cell 164(6):1233–1247. https://doi.org/10.1016/j.cell.2016.01.049
Palza H, Zapata PA, Angulo-Pineda C (2019) Electroactive smart polymers for biomedical applications. Materials. https://doi.org/10.3390/ma12020277
Pardi N, Muramatsu H, Weissman D, Karikó K (2013) In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol 969:29–42. https://doi.org/10.1007/978-1-62703-260-5_2
Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D (2015) Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release 217:345–351. https://doi.org/10.1016/j.jconrel.2015.08.007
Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, Wagner W, Granados A, Greenhouse J, Walker M, Willis E, Yu JS, McGee CE, Sempowski GD, Mui BL, Tam YK, Huang YJ, Vanlandingham D, Holmes VM, Balachandran H, Sahu S, Lifton M, Higgs S, Hensley SE, Madden TD, Hope MJ, Karikó K, Santra S, Graham BS, Lewis MG, Pierson TC, Haynes BF, Weissman D (2017) Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543(7644):248–251. https://doi.org/10.1038/nature21428
Pardi N, Hogan MJ, Porter FW, Weissman D (2018) mRNA vaccines – a new era in vaccinology. Nat Rev Drug Discov 17(4):261–279. https://doi.org/10.1038/nrd.2017.243
Parent A, Bisaillon M (2006) Synergy between transcription and mRNA processing events. Med Sci 22(6–7):626–632. https://doi.org/10.1051/medsci/20062267626 (SynergieentrelescomplexesdetranscriptionetdematurationdesARNmessagers)
Pascolo S (2004) Messenger RNA-based vaccines. Expert Opin Biol Ther 4(8):1285–1294. https://doi.org/10.1517/14712598.4.8.1285
Pawar GN, Parayath NN, Nocera AL, Bleier BS, Amiji MM (2018) Direct CNS delivery of proteins using thermosensitive liposome-in-gel carrier by heterotopic mucosal engrafting. PLoS ONE 13(12):e0208122. https://doi.org/10.1371/journal.pone.0208122
Peng J, Schoenberg DR (2005) mRNA with a <20-nt poly(A) tail imparted by the poly(A)-limiting element is translated as efficiently in vivo as long poly(A) mRNA. RNA 11(7):1131–1140. https://doi.org/10.1261/rna.2470905
Perche F, Benvegnu T, Berchel M, Lebegue L, Pichon C, Jaffrès PA, Midoux P (2011) Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 7(4):445–453. https://doi.org/10.1016/j.nano.2010.12.010
Perche F, Clemençon R, Schulze K, Ebensen T, Guzmán CA, Pichon C (2019) Neutral lipopolyplexes for in vivo delivery of conventional and replicative RNA vaccine. Mole Ther Nucl Acids 17:767–775. https://doi.org/10.1016/j.omtn.2019.07.014
Petsch B, Schnee M, Vogel AB, Lange E, Hoffmann B, Voss D, Schlake T, Thess A, Kallen KJ, Stitz L, Kramps T (2012) Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol 30(12):1210–1216. https://doi.org/10.1038/nbt.2436
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC (2020) Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med 383(27):2603–2615. https://doi.org/10.1056/NEJMoa2034577
Pollard C, De Koker S, Saelens X, Vanham G, Grooten J (2013) Challenges and advances towards the rational design of mRNA vaccines. Trends Mol Med 19(12):705–713. https://doi.org/10.1016/j.molmed.2013.09.002
Proudfoot N (2004) New perspectives on connecting messenger RNA 3’ end formation to transcription. Curr Opin Cell Biol 16(3):272–278. https://doi.org/10.1016/j.ceb.2004.03.007
Ramamoorth M, Narvekar A (2015) Non viral vectors in gene therapy–An overview. J Clin Diagn Res 9(1):Ge0-06. https://doi.org/10.7860/jcdr/2015/10443.5394
Ramasamy T, Ruttala HB, Gupta B, Poudel BK, Choi HG, Yong CS, Kim JO (2017) Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release 258:226–253. https://doi.org/10.1016/j.jconrel.2017.04.043
Ramasamy T, Ruttala HB, Kaliraj K, Poudel K, Jin SG, Choi H-G, Ku SK, Yong CS, Kim JO (2019) Polypeptide derivative of metformin with the combined advantage of a gene carrier and anticancer activity. ACS Biomater Sci Eng 5(10):5159–5168. https://doi.org/10.1021/acsbiomaterials.9b00982
Ramasamy T, Munusamy S, Ruttala HB, Kim JO (2021) Smart nanocarriers for the delivery of nucleic acid-based therapeutics: a comprehensive review. Biotechnol J 16(2):e1900408. https://doi.org/10.1002/biot.201900408
Redzic JS, Balaj L, van der Vos KE, Breakefield XO (2014) Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol 28:14–23. https://doi.org/10.1016/j.semcancer.2014.04.010
Rejman J, Tavernier G, Bavarsad N, Demeester J, De Smedt SC (2010) mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J Control Release 147(3):385–391. https://doi.org/10.1016/j.jconrel.2010.07.124
Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS (2017) Modified mRNA vaccines protect against zika virus infection. Cell 168(6):1114-1125. e1110. https://doi.org/10.1016/j.cell.2017.02.017
Ruttala HB, Ramasamy T, Madeshwaran T, Hiep TT, Kandasamy U, Oh KT, Choi H-G, Yong CS, Kim JO (2018) Emerging potential of stimulus-responsive nanosized anticancer drug delivery systems for systemic applications. Arch Pharmacal Res 41(2):111–129. https://doi.org/10.1007/s12272-017-0995-x
Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, Lynn A, Bulychev A, McFadyen I, Chan J, Almarsson Ö, Stanton MG, Benenato KE (2018) A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther 26(6):1509–1519. https://doi.org/10.1016/j.ymthe.2018.03.010
Sahin U, Karikó K, Türeci Ö (2014) mRNA-based therapeutics — Developing a new class of drugs. Nat Rev Drug Discov 13(10):759–780. https://doi.org/10.1038/nrd4278
Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, Maurus D, Schwarck-Kokarakis D, Kuhn AN, Omokoko T, Kranz LM, Diken M, Kreiter S, Haas H, Attig S, Rae R, Cuk K, Kemmer-Brück A, Breitkreuz A, Tolliver C, Caspar J, Quinkhardt J, Hebich L, Stein M, Hohberger A, Vogler I, Liebig I, Renken S, Sikorski J, Leierer M, Müller V, Mitzel-Rink H, Miederer M, Huber C, Grabbe S, Utikal J, Pinter A, Kaufmann R, Hassel JC, Loquai C, Türeci Ö (2020) An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 585(7823):107–112. https://doi.org/10.1038/s41586-020-2537-9
Sasaki K, Sato Y, Okuda K, Iwakawa K, Harashima H (2022) mRNA-loaded lipid nanoparticles targeting dendritic cells for cancer immunotherapy. Pharmaceutics 14(8):1572. https://doi.org/10.3390/pharmaceutics14081572
Singh AP, Biswas A, Shukla A, Maiti P (2019) Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct Target Ther 4(1):33. https://doi.org/10.1038/s41392-019-0068-3
Sonenberg N, Gingras AC (1998) The mRNA 5’ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol 10(2):268–275. https://doi.org/10.1016/s0955-0674(98)80150-6
Sousa F, Dhaliwal HK, Gattacceca F, Sarmento B, Amiji MM (2019) Enhanced anti-angiogenic effects of bevacizumab in glioblastoma treatment upon intranasal administration in polymeric nanoparticles. J Control Release 309:37–47. https://doi.org/10.1016/j.jconrel.2019.07.033
Stacey DW, Allfrey VG (1976) Microinjection studies of duck globin messenger RNA translation in human and avian cells. Cell 9(4 pt 2):725–732. https://doi.org/10.1016/0092-8674(76)90136-7
Strenkowska M, Grzela R, Majewski M, Wnek K, Kowalska J, Lukaszewicz M, Zuberek J, Darzynkiewicz E, Kuhn AN, Sahin U, Jemielity J (2016) Cap analogs modified with 1, 2-dithiodiphosphate moiety protect mRNA from decapping and enhance its translational potential. Nucleic Acids Res 44(20):9578–9590. https://doi.org/10.1093/nar/gkw896
Su X, Fricke J, Kavanagh DG, Irvine DJ (2011) In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol Pharm 8(3):774–787. https://doi.org/10.1021/mp100390w
Sun Y, Sun Y, Zhao R, Gao K (2016) Intracellular delivery of messenger RNA by recombinant PP7 virus-like particles carrying low molecular weight protamine. BMC Biotechnol 16(1):46. https://doi.org/10.1186/s12896-016-0274-9
Sunshine JC, Peng DY, Green JJ (2012) Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm 9(11):3375–3383. https://doi.org/10.1021/mp3004176
Tavernier G, Andries O, Demeester J, Sanders NN, De Smedt SC, Rejman J (2011) mRNA as gene therapeutic: how to control protein expression. J Control Release 150(3):238–247. https://doi.org/10.1016/j.jconrel.2010.10.020
Thangam R, Paulmurugan R, Kang H (2022) Functionalized nanomaterials as tailored theranostic agents in brain imaging. Nanomaterials. https://doi.org/10.3390/nano12010018
Theofilopoulos AN, Baccala R, Beutler B, Kono DH (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23:307–336. https://doi.org/10.1146/annurev.immunol.23.021704.115843
Torchilin VP (2005) Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov 4(2):145–160. https://doi.org/10.1038/nrd1632
Uchida S, Kinoh H, Ishii T, Matsui A, Tockary TA, Takeda KM, Uchida H, Osada K, Itaka K, Kataoka K (2016) Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 82:221–228. https://doi.org/10.1016/j.biomaterials.2015.12.031
Udhayakumar VK, De Beuckelaer A, McCaffrey J, McCrudden CM, Kirschman JL, Vanover D, Van Hoecke L, Roose K, Deswarte K, De Geest BG, Lienenklaus S, Santangelo PJ, Grooten J, McCarthy HO, De Koker S (2017) Arginine-rich peptide-based mRNA nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic organization of the peptide. Adv Healthc Mater. https://doi.org/10.1002/adhm.201601412
Uzgün S, Akdemir O, Hasenpusch G, Maucksch C, Golas MM, Sander B, Stark H, Imker R, Lutz JF, Rudolph C (2010) Characterization of tailor-made copolymers of oligo(ethylene glycol) methyl ether methacrylate and N, N-dimethylaminoethyl methacrylate as nonviral gene transfer agents: Influence of macromolecular structure on gene vector particle properties and transfection efficiency. Biomacromol 11(1):39–50. https://doi.org/10.1021/bm9008759
Uzgün S, Nica G, Pfeifer C, Bosinco M, Michaelis K, Lutz JF, Schneider M, Rosenecker J, Rudolph C (2011) PEGylation improves nanoparticle formation and transfection efficiency of messenger RNA. Pharm Res 28(9):2223–2232. https://doi.org/10.1007/s11095-011-0464-z
Vaidyanathan S, Chen J, Orr BG, Banaszak Holl MM (2016a) Cationic polymer intercalation into the lipid membrane enables intact polyplex DNA escape from endosomes for gene delivery. Mol Pharm 13(6):1967–1978. https://doi.org/10.1021/acs.molpharmaceut.6b00139
Vaidyanathan S, Orr BG, Banaszak Holl MM (2016b) Role of cell membrane-vector interactions in successful gene delivery. Acc Chem Res 49(8):1486–1493. https://doi.org/10.1021/acs.accounts.6b00200
Verbeke R, Lentacker I, De Smedt SC, Dewitte H (2021) The dawn of mRNA vaccines: the COVID-19 case. J Control Release 333:511–520. https://doi.org/10.1016/j.jconrel.2021.03.043
Wang T, Upponi JR, Torchilin VP (2012) Design of multifunctional non-viral gene vectors to overcome physiological barriers: dilemmas and strategies. Int J Pharm 427(1):3–20. https://doi.org/10.1016/j.ijpharm.2011.07.013
Wang Y, Su HH, Yang Y, Hu Y, Zhang L, Blancafort P, Huang L (2013) Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther 21(2):358–367. https://doi.org/10.1038/mt.2012.250
Wang Y, Zhang R, Tang L, Yang L (2022) Nonviral delivery systems of mRNA vaccines for cancer gene therapy. Pharmaceutics. https://doi.org/10.3390/pharmaceutics14030512
Weissman D (2015) mRNA transcript therapy. Expert Rev Vaccines 14(2):265–281. https://doi.org/10.1586/14760584.2015.973859
Wickens M, Anderson P, Jackson RJ (1997) Life and death in the cytoplasm: messages from the 3’ end. Curr Opin Genet Dev 7(2):220–232. https://doi.org/10.1016/s0959-437x(97)80132-3
Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J (2015) Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol 33(8):870–876. https://doi.org/10.1038/nbt.3298
Wolfe MS (2014) Targeting mRNA for Alzheimer’s and related dementias. Scientifica 2014:757549. https://doi.org/10.1155/2014/757549
Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL (1990) Direct gene transfer into mouse muscle in vivo. Science 247(4949 Pt 1):1465–1468. https://doi.org/10.1126/science.1690918
Xiao Y, Chen J, Zhou H, Zeng X, Ruan Z, Pu Z, Jiang X, Matsui A, Zhu L, Amoozgar Z, Chen DS, Han X, Duda DG, Shi J (2022) Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat Commun 13(1):758. https://doi.org/10.1038/s41467-022-28279-8
Xiong Q, Lee GY, Ding J, Li W, Shi J (2018) Biomedical applications of mRNA nanomedicine. Nano Res 11(10):5281–5309. https://doi.org/10.1007/s12274-018-2146-1
Yadav S, Gattacceca F, Panicucci R, Amiji MM (2015) Comparative biodistribution and pharmacokinetic analysis of cyclosporine-A in the brain upon intranasal or intravenous administration in an oil-in-water nanoemulsion formulation. Mol Pharm 12(5):1523–1533. https://doi.org/10.1021/mp5008376
Yan Y, Xiong H, Zhang X, Cheng Q, Siegwart DJ (2017) Systemic mRNA delivery to the lungs by functional polyester-based carriers. Biomacromol 18(12):4307–4315. https://doi.org/10.1021/acs.biomac.7b01356
Yang J, Arya S, Lung P, Lin Q, Huang J, Li Q (2019) Hybrid nanovaccine for the co-delivery of the mRNA antigen and adjuvant. Nanoscale 11(45):21782–21789. https://doi.org/10.1039/C9NR05475H
Yasar H, Biehl A, De Rossi C, Koch M, Murgia X, Loretz B, Lehr C-M (2018) Kinetics of mRNA delivery and protein translation in dendritic cells using lipid-coated PLGA nanoparticles. J Nanobiotechnol 16(1):72. https://doi.org/10.1186/s12951-018-0401-y
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG (2014) Non-viral vectors for gene-based therapy. Nat Rev Genet 15(8):541–555. https://doi.org/10.1038/nrg3763
Zeyer F, Mothes B, Will C, Carevic M, Rottenberger J, Nürnberg B, Hartl D, Handgretinger R, Beer-Hammer S, Kormann MSD (2016) mRNA-mediated gene supplementation of toll-like receptors as treatment strategy for asthma in vivo. PLoS ONE 11(4):e0154001. https://doi.org/10.1371/journal.pone.0154001
Zhang Y, Pelet JM, Heller DA, Dong Y, Chen D, Gu Z, Joseph BJ, Wallas J, Anderson DG (2013) Lipid-modified aminoglycoside derivatives for in vivo siRNA delivery. Adv Mater 25(33):4641–4645. https://doi.org/10.1002/adma.201301917
Zhang R, Wu F, Wu L, Tian Y, Zhou B, Zhang X, Huang R, Yu C, He G, Yang L (2018) Novel self-assembled micelles based on cholesterol-modified antimicrobial peptide (DP7) for safe and effective systemic administration in animal models of bacterial infection. Antimicrob Agents Chemother. https://doi.org/10.1128/aac.00368-18
Zhang C, Zhang X, Zhao W, Zeng C, Li W, Li B, Luo X, Li J, Jiang J, Deng B, McComb DW, Dong Y (2019a) Chemotherapy drugs derived nanoparticles encapsulating mRNA encoding tumor suppressor proteins to treat triple-negative breast cancer. Nano Res 12(4):855–861. https://doi.org/10.1007/s12274-019-2308-9
Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, Holland EC, Stephan MT (2019b) Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun 10(1):3974. https://doi.org/10.1038/s41467-019-11911-5
Zhang X, Men K, Zhang Y, Zhang R, Yang L, Duan X (2019c) Local and systemic delivery of mRNA encoding survivin-T34A by lipoplex for efficient colon cancer gene therapy. Int J Nanomed 14:2733–2751. https://doi.org/10.2147/ijn.S198747
Zhang R, Tang L, Tian Y, Ji X, Hu Q, Zhou B, Ding Z, Xu H, Yang L (2020a) DP7-C-modified liposomes enhance immune responses and the antitumor effect of a neoantigen-based mRNA vaccine. J Control Release 328:210–221. https://doi.org/10.1016/j.jconrel.2020.08.023
Zhang R, Tang L, Tian Y, Ji X, Hu Q, Zhou B, Zhenyu D, Heng X, Yang L (2020b) Cholesterol-modified DP7 enhances the effect of individualized cancer immunotherapy based on neoantigens. Biomaterials 241:119852. https://doi.org/10.1016/j.biomaterials.2020.119852
Zhang D, Wang G, Yu X, Wei T, Farbiak L, Johnson LT, Taylor AM, Xu J, Hong Y, Zhu H, Siegwart DJ (2022a) Enhancing CRISPR/Cas gene editing through modulating cellular mechanical properties for cancer therapy. Nat Nanotechnol 17(7):777–787. https://doi.org/10.1038/s41565-022-01122-3
Zhang P, Li Y, Tang W, Zhao J, Jing L, McHugh KJ (2022b) Theranostic nanoparticles with disease-specific administration strategies. Nano Today 42:101335. https://doi.org/10.1016/j.nantod.2021.101335
Zhang Y, Hu Y, Tian H, Chen X (2022c) Opportunities and challenges for mRNA delivery nanoplatforms. J Phys Chem Lett 13(5):1314–1322. https://doi.org/10.1021/acs.jpclett.1c03898
Zhitnyuk Y, Gee P, Lung MSY, Sasakawa N, Xu H, Saito H, Hotta A (2018) Efficient mRNA delivery system utilizing chimeric VSVG-L7Ae virus-like particles. Biochem Biophys Res Commun 505(4):1097–1102. https://doi.org/10.1016/j.bbrc.2018.09.113
Zhong Z, Mc Cafferty S, Combes F, Huysmans H, De Temmerman J, Gitsels A, Vanrompay D, Portela Catani J, Sanders NN (2018) mRNA therapeutics deliver a hopeful message. Nano Today 23:16–39. https://doi.org/10.1016/j.nantod.2018.10.005
Zhu Y, Zhu L, Wang X, Jin H (2022) RNA-based therapeutics: an overview and prospectus. Cell Death Dis 13(7):644. https://doi.org/10.1038/s41419-022-05075-2
Zohra FT, Chowdhury EH, Tada S, Hoshiba T, Akaike T (2007) Effective delivery with enhanced translational activity synergistically accelerates mRNA-based transfection. Biochem Biophys Res Commun 358(1):373–378. https://doi.org/10.1016/j.bbrc.2007.04.059
Zohra FT, Chowdhury EH, Akaike T (2009) High performance mRNA transfection through carbonate apatite-cationic liposome conjugates. Biomaterials 30(23–24):4006–4013. https://doi.org/10.1016/j.biomaterials.2009.02.050
Zohra FT, Maitani Y, Akaike T (2012) mRNA delivery through fibronectin associated liposome-apatite particles: a new approach for enhanced mRNA transfection to mammalian cell. Biol Pharm Bull 35(1):111–115. https://doi.org/10.1248/bpb.35.111
Acknowledgements
This project was financially supported by a grant from the Ministry of Higher Education, Malaysia (MoHE) FRGS/1/2018/STG05/MUSM/02/3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Karim, M.E., Haque, S.T., Al-Busaidi, H. et al. Scope and challenges of nanoparticle-based mRNA delivery in cancer treatment. Arch. Pharm. Res. 45, 865–893 (2022). https://doi.org/10.1007/s12272-022-01418-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01418-x