
Abstract
Pathogenic factors associated with maternal hypercholesterolemia, obesity, and diabetic conditions during pregnancy influence fetal development and predispose offspring to cardiovascular disease. Animal models have established cause–effect relationships consistent with epidemiological findings in humans and have demonstrated, in principle, that interventions before or during pregnancy can reduce or prevent pathogenic in utero programming. However, little is known about the mechanisms by which maternal dysmetabolic conditions enhance disease susceptibility in offspring. Identification of these mechanisms is rendered more difficult by the fact that programming effects in offspring may be latent and may require conventional risk factors and inherited genetic co-factors to become clinically manifest. Given the increasing prevalence of maternal risk factors, which is expected to lead to a wave of cardiovascular disease in the coming decades, and the length of prospective studies on developmental programming in humans, greater-than-usual emphasis on experimental models and translational studies is necessary.
Similar content being viewed by others


Introduction
The impact of gender on cardiovascular disease is well established and it would nowadays be inconceivable to carry out epidemiological studies on pathogenic factors or clinical intervention trials without separate analysis of female and male subjects. Unfortunately, identification of gender differences at the metabolic, endocrine, and cellular level has not yet translated into the anticipated clinical benefits, as shown by the failure of late-onset estrogen replacement to protect postmenopausal women against cardiovascular disease [1–3]. Yet, in addition to differences in pathogenetic mechanisms and responses to drugs there is a third, far less recognized area in which a greater focus on females promises substantial benefits.
Extensive epidemiological evidence supports the concept that the in utero environment influences the susceptibility to many diseases later in life. Barker and colleagues first noticed an association between low birthweight and adult cardiovascular disease, hypertension, and diabetes [4–6]. Later studies refined this by showing that adult cardiovascular risk was associated not with low birthweight per se, which may also reflect premature birth, but with impaired intrauterine growth [7]. However, growth retardation is an outcome parameter of fetal development which may result from many pathogenetically diverse causes, ranging from extreme maternal undernutrition to mechanical obstruction of the uterine artery. In addition, fetal growth, especially in experimental models, may also vary considerably due to sibling competition. It is therefore increasingly recognized that elucidation of developmental programming should focus on maternal pathogenic factors, rather than outcome parameters [8]. Furthermore, maternal conditions associated with overnutrition, such as hypercholesterolemia, obesity, hypertension, insulin resistance, and type 2 diabetes are far more prevalent in the developed world than severe undernutrition [9]. The increasing prevalence of maternal obesity, gestational diabetes, and other dysmetabolic conditions, their association with offspring obesity and diabetic conditions, and the mounting evidence for atherogenic programming by specific maternal factors in experimental models suggest that pathogenic in utero programming may contribute to the expected wave of cardiovascular disease in their offspring [9–18].
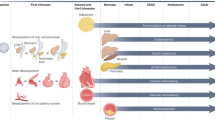
On the other hand, results in experimental models suggest that pathogenic in utero programming may be prevented by relatively simple and safe interventions, as well as by treatment of the underlying maternal condition. In addition, preventive measures are likely to yield lifelong benefits for offspring. The following will provide a brief overview of developmental programming from a translational perspective (Fig. 1).

Fetal programming of cardiovascular disease. Modified from reference [87]
Programming by Maternal Undernutrition
The fact that so many different factors may lead to impaired fetal growth in humans is a major obstacle in the search for the programming mechanisms, in particular when data from different continents with obvious genetic and dietary differences are compared. One way to reduce confounders is to focus on relatively homogeneous populations with a clearly defined and temporally limited cause of growth retardation, such as maternal starvation for children born during the Dutch Hunger Winter (1944–1945) [19]. Although still ongoing, studies of this population have already shown a greater prevalence of obesity and, in some subgroups, an increased insulin resistance. Animal models of extreme undernutrition have established that exposure to excessive maternal glucocorticoids (or exogenously administered dexamethasone) during late pregnancy plays an important role in fetal growth retardation, as well as in adult insulin hyperglycemia, hyperinsulinemia [20, 21], hypertension [22, 23], and atherogenesis [24]. Maternal protein restriction also induced offspring hypertension in rats [25, 26].
However, other studies in prematurely born human infants have indicated that increased, rather than reduced, growth during a critical postnatal period promotes insulin resistance [27]. This points to a major practical problem. In contrast to conventional early predictors of adult cardiovascular risk, such as childhood endothelial dysfunction, atherogenesis, obesity, and insulin resistance, the pathogenic effects of fetal growth retardation cannot be corrected by compensatory weight gain after birth. Indeed, accelerated growth during childhood only increases later insulin resistance and hypertension, particularly in prematurely born children [28].
Studies of fetal undernutrition have also prompted the hypothesis that fetal programming represents an attempt of the fetus to adapt to adverse conditions encountered in utero. If similar conditions, i.e. undernutrition, prevail later in life, such adaptation would be beneficial, whereas it would be detrimental in case of normal or abundant nutrition [29]. If this hypothesis is correct, a therapeutical corollary would be that low-caloric diets should prevent cardiovascular disease in subjects born underweight for their gestational age. This corollary is at least consistent with the observation that postnatal overnutrition is particularly pathogenic in these subjects. The opposite corollary, i.e., that programming by gestational hypercholesterolemia, insulin resistance, or diabetes should be least pathogenic under continued postnatal exposure to conventional risk factors of these conditions seems distinctly less attractive.
Thus, although the correlation between fetal growth retardation and adult disease is supported by the majority of epidemiological studies and some animal studies, it is unlikely that pathogenic programming associated with impaired fetal growth will be preventable, unless growth retardation itself is prevented. In contrast, other fields of developmental programming have made significant progress towards this goal, as outlined below.
Programming by Maternal Hypercholesterolemia, Obesity, and Diabetic Conditions
Although clinical manifestations of atherosclerosis typically occur in adults, atherosclerotic lesions are found in most children [30–32] and prodromal lesions, fatty streaks, may already be formed during fetal development. The first indication that in utero exposure to cholesterol may program adult cardiovascular disease came from the observation that maternal hypercholesterolemia, even if temporary and limited to pregnancy only, is associated with a marked increase in fatty streaks in the aorta of premature human fetuses [16]. The same study also showed that fetal cholesterol levels at the end of the second trimester are very high, even in fetuses of normocholesterolemic mothers, and decline linearly until term birth. This is consistent with the well-documented fact that in the absence of inherited genetic defects of lipid metabolism, such as familial hypercholesterolemia, cholesterol levels of term-born children are very low, even if their mothers are extensively hypercholesterolemic. It was therefore conceivable that fetal lesions would regress towards full term. Instead, the Fate of Early Lesions in Children (FELIC) study, a morphometric assessment of aortic atherosclerosis in normocholesterolemic children who died before the age of 14, demonstrated that maternal hypercholesterolemia is associated with greater atherogenesis throughout childhood [17]. This observation could not be explained by conventional risk factors in mothers or children and suggested programming by maternal hypercholesterolemia or the ensuing increase in oxidative stress. A retrospective study correlating maternal cholesterol levels with the severity of myocardial infarction in young adults is currently under way to establish the effect of fetal programming on clinical manifestations of atherosclerosis.
In the human subjects of the FELIC study, genetic traits inherited from hypercholesterolemic mothers are likely to have contributed to increased atherosclerosis in their offspring. Direct experimental evidence for the causal role of maternal hypercholesterolemia was obtained in genetically more uniform models. For example, temporary, diet-induced maternal hypercholesterolemia prior to and during pregnancy led to dose-dependent increases in fetal lesion formation [33] and postnatal atherogenesis in NZW rabbits [34]. Conversely, maternal cholesterol-lowering with cholestyramine, considered safe during pregnancy, or with a natural antioxidant, vitamin E, greatly reduced offspring atherosclerosis [33, 34]. Similarly, maternal treatment with a statin during pregnancy (contraindicated in humans) reduced atherogenic programming in mice [35]. The protective effect of antioxidants and cholesterol-lowering drugs also indicated an important role of oxidative stress in developmental programming, which is discussed in the following chapter.
Mouse and rat models also provided valuable insights into fetal programming. For example, moderate maternal hypercholesterolemia led to persistent changes in gene expression in the arterial intima and media prior to histologically detectable atherogenesis [36]. It also enhanced offspring atherosclerosis in apoE-deficient (ApoE−/−) mice [37], although others had failed to see an effect in this strain [38]. One of the most important results was obtained by a comparison of heterozygous offspring generated by crossing wild-type apoE+/+ females with apoE−/− males, and vice versa. Thus, all offspring were genetically identical, but exposed to either normal or mildly hypercholesterolemic conditions in utero, depending on their mothers [39]. The degree of hypercholesterolemia achievable in apoE+/− offspring was insufficient to induce significant atherosclerosis, but pathogenic programming manifested itself in the form of dramatically increased carotid atherosclerosis, once an additional atherogenic stimulus was added, in this case a non-constricting carotid cuff [39]. These results highlight the fact that fetal programming effects may be latent and become apparent only in the presence of additional or more severe conventional risk factors, or, as shown recently in a model of spontaneous stroke, a combination of inherited genetic susceptibility and dietary factors acting synergistically [40, 41].
Wild-type rat models do not usually develop sufficiently high plasma cholesterol levels to cause atherosclerosis, but have indicated other mechanisms by which maternal high-fat, high-cholesterol diets during pregnancy may promote cardiovascular disease, such as impaired vascular reactivity in offspring [42–45]. Some of these effects were enhanced by maternal diabetes [42], and conversely, high-fat diets promoted insulin resistance in offspring [46]. Maternal exposure of rats to a high carbohydrate diet resulted in similar programming of hyperinsulinemia, which could be reduced by low-carbohydrate diets [47].
Murine models of obesity and gestational diabetes have identified programming of factors involved in insulin signaling and insulin resistance itself [48]. However, caution is indicated when extrapolating the effects of high-fat diets and obesity in mice to humans, because the C57\BL6 strains appears to be genetically predisposed to type 2 diabetes [49, 50]. Furthermore, in many rodent models a clear distinction between pathogenic factors is impossible, because standard high-fat, high-cholesterol diets induce not just hypercholesterolemia but also obesity and insulin resistance [51]. Such a distinction is less important for translational purposes, though, because obesity, insulin resistance and combined hyperlipidemia are prevalent in many human mothers, and synergism of pathogenic mechanism programmed in utero is likely.
Putative Mechanisms of Early-life Programming
Hypercholesterolemia is associated with enhanced lipid peroxidation and a wide range of oxidized fatty acids and other markers of lipid peroxidation are increased in fetal plasma and aortic lesions [16, 52–54]. The first indication that increased oxidative stress might play a role in atherogenic programming was provided by a study in premature human fetuses which showed that intracranial arteries, which had higher activities of antioxidant enzymes, developed less atherosclerosis in response to maternal hypercholesterolemia than extracranial arteries [52]. A similar observation later linked an acceleration of intracranial atherogenesis in elderly human subject to an age-related decrease in antioxidative protection [55]. Given that OxLDL enhances adult atherogenesis by a number of mechanisms, including interference with many oxidation-sensitive nuclear signaling pathways [56], we proposed that oxidative stress is important in developmental programming by maternal hypercholesterolemia [17, 57]. This notion was supported by the prevention of atherogenic programming achieved by maternal treatments which reduced lipid peroxidation either directly (antioxidants) or by reducing plasma cholesterol (cholestyramine, statins) [33, 34]. Later studies also established an involvement of oxidative stress in the fetal programming of vascular dysfunction and hypertension [58].
Lipid peroxidation products (reactive aldehydes, ketones, and oxidized phospholipids) modify biological properties of many proteins by forming adducts with free amino groups. They may also affect DNA. In fact, much attention has focused on DNA methylation and other epigenetic effects [59]. In utero conditions indeed result in specific epigenetic changes [60, 61]. However, there is no evidence that epigenetic changes actually cause or contribute to increased disease manifestation in offspring. Another possibly related mechanism is the alteration of mitochondrial DNA [46]. This is intuitively attractive, because mitochondrial DNA is inherited exclusively from the mother and would therefore allow the transfer of fetal programming “memory” to successive generations.
Finally, we have recently shown in both rabbit and murine models that immune mechanisms may influence fetal programming. Maternal immunization with homologous OxLDL prior to hypercholesterolemic pregnancy effectively reduced atherogenic programming in their offspring, probably by reducing fetal exposure to circulating mildly oxidized LDL [18]. In fact, increased formation of antibodies to oxidation-specific epitopes had previously been shown to reduce atherogenesis in adult animals, in part by formation and rapid elimination of immune complexes [62]. Unexpectedly, the beneficial effect was not limited to offspring of hypercholesterolemic mothers. Furthermore, titers of protective IgM antibodies and IgM-LDL immune complexes were persistently increased in adult offspring of immunized mothers. Naïve adult offspring never exposed to increased oxidative stress also showed markedly increased B cell-dependent IgM and IgG responses to antigenic challenge with selective oxidation-specific epitopes. These results demonstrate that immature fetal lymphocytes are programmed in utero. Previously, it was presumed that maternal adaptive immunity protects the neonate mainly by transplacental passage of maternal IgG, and in utero programming of B and T cells had only be shown in connection with increased IgE responses to allergens. Our findings showed that it is possible to modulate in utero programming of beneficial IgM and IgG responses by maternal immunization prior to pregnancy.
Elucidation of fetal immune programming is still in its infancy, but the above data also emphasize the importance of inflammation in in utero programming. Whether its role will be as complex as in atherosclerosis remains to be seen [63]. All of the maternal conditions of overnutrition discussed above promote inflammation. Furthermore, markers of inflammation, such as CRP, correlate with the extent of fetal atherogenesis in humans [64]. Similarly, increased CRP was observed in infants of mothers with type 1 diabetes [65]. Thus, proinflammatory cytokines will have to be considered as potential causes of programming, and makers of inflammation as potential predictors.
A good example that fetal programming by the same maternal factor may involve not just one but many of the mechanisms described above is provided by maternal smoking. This has been shown to increase formation of reactive oxygen species or reduce antioxidant activities [66, 67], to affect immune mechanisms [68], to impair the development of the lung and heart [69, 70], to affect mitochondrial DNA [67], and to enhance hypertension [71] in human and animal offspring.
The Role of the Placenta
The atherogenic programming effect of maternal hypercholesterolemia and the observation of strikingly high fetal cholesterol levels at the end of second trimester [16] raised new interest in maternal–fetal cholesterol transport. Could it be that maternal cholesterol crosses the placental barrier, which is impenetrable for lipoprotein particles? This possibility had already been postulated because of the survival of human fetuses with the Smith Lemli Opitz syndrome, which cannot synthesize cholesterol and therefore depend on maternal cholesterol [72]. Evidence for maternal–fetal cholesterol transport had also been provided by the identification of active transport mechanisms on the maternal side of the placenta in rabbits [73] and hamsters [74]. The last step in transplacental cholesterol transport mechanism—the export from endothelial cells lining the placental villi into the lumen of fetal microvessels—has recently been elucidated [75]. Results indicate that regulation of these mechanisms by increased fetal demand for cholesterol differs from that induced by maternal hypercholesterolemia [76]. Increased cholesterol transport, at least during parts of gestation, may therefore constitute one mechanism by which maternal hypercholesterolemia affects fetal programming.
In addition to maternal factors crossing the placental barrier, it is important to remember that the placenta is not a passive filter between mother and fetus, but that it can be both target and source of pathogenic factors affecting the fetus. A recent comparison of the activities of antioxidant enzymes in human maternal and fetal plasma and placental tissue from normo- and hypercholesterolemic subjects has shown that the placenta can both protect against, and contribute to, fetal oxidative stress [77]. Unfortunately, the placenta of many lower animal models shows significant anatomical and functional differences, compared to the human placenta, which complicates the investigation of its role in developmental programming.
Programming of Hypertension
Hypertension is a major risk factor of cardiovascular disease that is influenced by early-life programming, and hypertension during pregnancy is a leading cause of maternal and perinatal mortality. However, there is little evidence that maternal hypertension directly affects fetal programming. In fact, most insights into in utero modulation of hypertensive mechanisms were obtained in models of maternal hypercholesterolemia or obesity [42, 44, 78, 79] or impaired fetal growth [22, 80, 81]. Nevertheless, fetal exposure to high salt concentrations promotes offspring hypertension in both salt-sensitive and salt-resistant rat strains [82, 83]. A more complex causality was recently established in a novel rat model combining genetic salt susceptibility, expression of human cholesterol ester transfer protein, and mild dietary salt exposure [40]. Salt exposure began during pregnancy, at weaning, or early adult age. Both fetal and at weaning onset of salt exposure resulted in extensive spontaneous stroke. Thus, the critical period was not limited to fetal development, but included lactation and infancy [40, 41].
A Word of Caution
Throughout this review, the authors have taken some liberty with the terms referring to developmental programming. This reflects the present lack of understanding not only of the mechanisms involved, but also of the timeframe. Fetal (i.e., in utero) programming and programming during lactation probably constitute a single entity. Childhood conditions also markedly influence adult disease manifestation and may enhance or attenuate the effects of fetal programming. Again, the mechanisms are largely unknown, but conditioning during childhood clearly represents a different entity than fetal programming. Finally, we should be aware that early-life exposure to pathogenic factors may predispose one to adult disease manifestation by at least three very different mechanisms. First, by actual programming events resulting in permanent differences in gene expression or epigenetic changes. Second, by impaired development of tissues or organs, which may persist over time in the form of altered cellular composition or reduced functionality. The latter is probably true for the lungs of asthmatic children whose mothers smoked during pregnancy. Third, neonates/infants may just be particularly vulnerable to specific injury, e.g. by toxins or free radicals. In this case, the same insult would cause lesser damage in adults, but the mechanisms would not be different, and no programming would be involved. From a translational perspective, this distinction should not be very relevant, as long as we can find ways to prevent the pathogenic events in the fetus from occurring.
Conclusion
Increasing evidence indicates that maternal hypercholesterolemia, obesity, and diabetic conditions during pregnancy influence fetal development and predispose offspring to cardiovascular disease. Animal models have established cause–effect relationships for some maternal factors and led to the identification of specific epigenetic changes, but the nature of fetal programming and the mechanisms by which they actually promote adult disease remain largely unknown. Similarly, most of the maternal factors responsible for fetal programming remain unknown. The National Children's Study, a prospective study designed to document maternal pathologies and exposure to environmental risk factors and to follow 100,000 of their children to the age of 25 years, will probably identify many more of these factors and establish better correlations with offspring effects [84–86]. Sadly, this will come rather late for the children of the increasing number of current high-risk mothers. A greater-than-usual emphasis on experimental models and on translational studies is indicated in order to explore the mechanisms and to identify potential targets for intervention [87].
References
Grodstein, F., Manson, J. E., Colditz, G. A., Willett, W. C., Speizer, F. E., & Stampfer, M. J. (2000). A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Annals of Internal Medicine, 133(12), 933–941.
Manson, J. E., Hsia, J., Johnson, K. C., Rossouw, J. E., Assaf, A. R., Lasser, N. L., et al. (2003). Estrogen plus progestin and the risk of coronary heart disease. New England Journal of Medicine, 349(6), 523–534.
Vandenbroucke, J. P. (2009). The HRT controversy: observational studies and RCTs fall in line. Lancet, 373(9671), 1233–1235.
Barker, D. J., Winter, P. D., Osmond, C., Margetts, B., & Simmonds, S. J. (1989). Weight in infancy and death from ischaemic heart disease. Lancet, 2(8663), 577–580.
Martyn, C. N., Gale, C. R., Jespersen, S., & Sherriff, S. B. (1998). Impaired fetal growth and atherosclerosis of carotid and peripheral arteries. Lancet, 352(9123), 173–178.
Barker, D. J. (2007). The origins of the developmental origins theory. Journal of Internal Medicine, 261(5), 412–417.
Kaijser, M., Bonamy, A. K., Akre, O., Cnattingius, S., Granath, F., Norman, M., et al. (2008). Perinatal risk factors for ischemic heart disease: disentangling the roles of birth weight and preterm birth. Circulation, 117(3), 405–410.
Gillman, M. W. (2002). Epidemiological challenges in studying the fetal origins of adult chronic disease. International Journal of Epidemiology, 31(2), 294–299.
Hedley, A. A., Ogden, C. L., Johnson, C. L., Carroll, M. D., Curtin, L. R., & Flegal, K. M. (2004). Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA, 291(23), 2847–2850.
Dabelea, D., Hanson, R. L., Lindsay, R. S., Pettitt, D. J., Imperatore, G., Gabir, M. M., et al. (2000). Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes, 49(12), 2208–2211.
Gillman, M. W., Rifas-Shiman, S., Berkey, C. S., Field, A. E., & Colditz, G. A. (2003). Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics, 111(3), e221–e226.
Dabelea, D. (2007). The predisposition to obesity and diabetes in offspring of diabetic mothers. Diabetes Care, 30(Suppl 2), S169–S174.
Clausen, T. D., Mathiesen, E. R., Hansen, T., Pedersen, O., Jensen, D. M., Lauenborg, J., et al. (2008). High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: the role of intrauterine hyperglycemia. Diabetes Care, 31(2), 340–346.
Wright, C. S., Rifas-Shiman, S. L., Rich-Edwards, J. W., Taveras, E. M., Gillman, M. W., & Oken, E. (2009). Intrauterine exposure to gestational diabetes, child adiposity, and blood pressure. American Journal of Hypertension, 22(2), 215–220.
Oken, E., Levitan, E. B., & Gillman, M. W. (2008). Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis. International Journal of Obesity (London), 32(2), 201–210.
Napoli, C., D'Armiento, F. P., Mancini, F. P., Postiglione, A., Witztum, J. L., Palumbo, G., & Palinski, W. (1997). Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. Journal of Clinical Investigation, 100(11), 2680–2690.
Napoli, C., Glass, C. K., Witztum, J. L., Deutsch, R., D'Armiento, F. P., & Palinski, W. (1999). Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet, 354(9186), 1234–1241.
Yamashita, T., Freigang, S., Eberle, C., Pattison, J., Gupta, S., Napoli, C., et al. (2006). Maternal immunization programs postnatal immune responses and reduces atherosclerosis in offspring. Circulation Research, 99(7), E51–E64.
Lumey, L. H., Stein, A. D., Kahn, H. S., van der Pal-de Bruin, K. M., Blauw, G. J., Zybert, P. A., et al. (2007). Cohort profile: the Dutch Hunger Winter families study. International Journal of Epidemiology, 36(6), 1196–1204.
Nyirenda, M. J., Lindsay, R. S., Kenyon, C. J., Burchell, A., & Seckl, J. R. (1998). Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. Journal of Clinical Investigation, 101(10), 2174–2181.
Moss, T. J., Sloboda, D. M., Gurrin, L. C., Harding, R., Challis, J. R., & Newnham, J. P. (2001). Programming effects in sheep of prenatal growth restriction and glucocorticoid exposure. American Journal of Physiology: Regulatory, Integrative & Comparative Physiology, 281(3), R960–R970.
Lindsay, R. S., Lindsay, R. M., Edwards, C. R., & Seckl, J. R. (1996). Inhibition of 11-beta-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension, 27(6), 1200–1204.
Aida, K., Wang, X. L., Wang, J., Li, C., McDonald, T. J., & Nathanielsz, P. W. (2004). Effect of betamethasone administration to the pregnant baboon at 0.75 gestation on placental eNOS distribution and activity. Placenta, 25(10), 780–787.
Yates, Z., Tarling, E.J., Langley-Evans, S.C. & Salter, A.M. (2008). Maternal undernutrition programmes atherosclerosis in the ApoE*3-Leiden mouse. British Journal of Nutrition1-10.
Brawley, L., Itoh, S., Torrens, C., Barker, A., Bertram, C., Poston, L., et al. (2003). Dietary protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatric Research, 54(1), 83–90.
Langley, S. C., & Jackson, A. A. (1994). Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clinical Science (London), 86(2), 217–222.
Singhal, A., Fewtrell, M., Cole, T. J., & Lucas, A. (2003). Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet, 361(9363), 1089–1097.
Barker, D. J., Osmond, C., Forsen, T. J., Kajantie, E., & Eriksson, J. G. (2005). Trajectories of growth among children who have coronary events as adults. New England Journal of Medicine, 353(17), 1802–1809.
Gluckman, P. D., & Hanson, M. A. (2004). Living with the past: evolution, development, and patterns of disease. Science, 305(5691), 1733–1736.
Stary, H. C. (1989). Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis, 9(1 Suppl), I19–I32.
Berenson, G. S., Wattigney, W. A., Tracy, R. E., Newman, W. P., III, Srinivasan, S. R., Webber, L. S., et al. (1992). Atherosclerosis of the aorta and coronary arteries and cardiovascular risk factors in persons aged 6 to 30 years and studied at necropsy (The Bogalusa Heart Study). American Journal of Cardiology, 70(9), 851–858.
Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. (1993). Natural history of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY Study. Arteriosclerosis & Thrombosis, 13(9), 1291–1298.
Napoli, C., Witztum, J. L., Calara, F., de Nigris, F., & Palinski, W. (2000). Maternal hypercholesterolemia enhances atherogenesis in normocholesterolemic rabbits, which is inhibited by antioxidant or lipid-lowering intervention during pregnancy: an experimental model of atherogenic mechanisms in human fetuses. Circulation Research, 87(10), 946–952.
Palinski, W., D'Armiento, F. P., Witztum, J. L., de Nigris, F., Casanada, F., Condorelli, M., et al. (2001). Maternal hypercholesterolemia and treatment during pregnancy influence the long-term progression of atherosclerosis in offspring of rabbits. Circulation Research, 89(11), 991–996.
Elahi, M. M., Cagampang, F. R., Anthony, F. W., Curzen, N., Ohri, S. K., & Hanson, M. A. (2008). Statin treatment in hypercholesterolemic pregnant mice reduces cardiovascular risk factors in their offspring. Hypertension, 51(4), 939–944.
Napoli, C., de Nigris, F., Welch, J. S., Calara, F. B., Stuart, R. O., Glass, C. K., & Palinski, W. (2002). Maternal hypercholesterolemia during pregnancy promotes early atherogenesis in LDL receptor-deficient mice and alters aortic gene expression determined by microarray. Circulation, 105(11), 1360–1367.
Goharkhay, N., Sbrana, E., Gamble, P. K., Tamayo, E. H., Betancourt, A., Villarreal, K., et al. (2007). Characterization of a murine model of fetal programming of atherosclerosis. American Journal of Obstetrics & Gynecology, 197(4), 416.e1–416.e5.
Madsen, C., Gnaes-Hansen, F., Moller, J., & Falk, E. (2003). Hypercholesterolemia in pregnant mice does not affect atherosclerosis in adult offspring. Atherosclerosis, 168(2), 221–228.
Alkemade, F. E., Gittenberger-de Groot, A. C., Schiel, A. E., VanMunsteren, J. C., Hogers, B., van Vliet, L. S., et al. (2007). Intrauterine exposure to maternal atherosclerotic risk factors increases the susceptibility to atherosclerosis in adult life. Arteriosclerosis, Thrombosis & Vascular Biology, 27(10), 2228–2235.
Decano, J. L., Viereck, J. C., McKee, A. C., Hamilton, J. A., Ruiz-Opazo, N., & Herrera, V. L. (2009). Early-life sodium exposure unmasks susceptibility to stroke in hyperlipidemic, hypertensive heterozygous Tg25 rats transgenic for human cholesteryl ester transfer protein. Circulation, 119(11), 1501–1509.
Palinski, W. (2009). Sodium exposure induces stroke in a genetically susceptible model: new insights into early-life factors modulating adult disease. Circulation, 119(11), 1459–1462.
Koukkou, E., Ghosh, P., Lowy, C., & Poston, L. (1998). Offspring of normal and diabetic rats fed saturated fat in pregnancy demonstrate vascular dysfunction. Circulation, 98(25), 2899–2904.
Ghosh, P., Bitsanis, D., Ghebremeskel, K., Crawford, M. A., & Poston, L. (2001). Abnormal aortic fatty acid composition and small artery function in offspring of rats fed a high fat diet in pregnancy. Journal of Physiology, 533(Pt 3), 815–822.
Khan, I., Dekou, V., Hanson, M., Poston, L., & Taylor, P. (2004). Predictive adaptive responses to maternal high-fat diet prevent endothelial dysfunction but not hypertension in adult rat offspring. Circulation, 110(9), 1097–1102.
Khan, I. Y., Dekou, V., Douglas, G., Jensen, R., Hanson, M. A., Poston, L., et al. (2005). A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. American Journal of Physiology: Regulatory, Integrative & Comparative Physiology, 288(1), R127–R133.
Taylor, P. D., McConnell, J., Khan, I. Y., Holemans, K., Lawrence, K. M., Sare-Anane, H., et al. (2005). Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. American Journal of Physiology: Regulatory, Integrative & Comparative Physiology, 288(1), R134–R139.
Srinivasan, M., Aalinkeel, R., Song, F., Mitrani, P., Pandya, J. D., Strutt, B., et al. (2006). Maternal hyperinsulinemia predisposes rat fetuses for hyperinsulinemia, and adult-onset obesity and maternal mild food restriction reverses this phenotype. American Journal of Physiology—Endocrinology & Metabolism, 290(1), E129–E134.
Yamashita, H., Shao, J., Qiao, L., Pagliassotti, M., & Friedman, J. E. (2003). Effect of spontaneous gestational diabetes on fetal and postnatal hepatic insulin resistance in Lepr(db/+) mice. Pediatric Research, 53(3), 411–418.
Surwit, R. S., Kuhn, C. M., Cochrane, C., McCubbin, J. A., & Feinglos, M. N. (1988). Diet-induced type II diabetes in C57BL/6J mice. Diabetes, 37(9), 1163–1167.
Surwit, R. S., Wang, S., Petro, A. E., Sanchis, D., Raimbault, S., Ricquier, D., et al. (1998). Diet-induced changes in uncoupling proteins in obesity-prone and obesity-resistant strains of mice. Proceedings of the National Academy of Sciences of the United States of America, 95(7), 4061–4065.
Merat, S., Casanada, F., Sutphin, M., Palinski, W., & Reaven, P. D. (1999). Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fru. Arteriosclerosis, Thrombosis, & Vascular Biology, 19(5), 1223–1230.
Napoli, C., Witztum, J. L., de Nigris, F., Palumbo, G., D'Armiento, F. P., & Palinski, W. (1999). Intracranial arteries of human fetuses are more resistant to hypercholesterolemia-induced fatty streak formation than extracranial arteries. Circulation, 99(15), 2003–2010.
Palinski, W., & Napoli, C. (1999). Pathophysiological events during pregnancy influence the development of atherosclerosis in humans. Trends in Cardiovascular Medicine, 9(7), 205–214.
Napoli, C., & Palinski, W. (2001). Maternal hypercholesterolemia during pregnancy influences the later development of atherosclerosis: clinical and pathogenic implications. European Heart Journal, 22(1), 4–9.
D'Armiento, F. P., Bianchi, A., de Nigris, F., Capuzzi, D. M., D'Armiento, M. R., Crimi, G., et al. (2001). Age-related effects on atherogenesis and scavenger enzymes of intracranial and extracranial arteries in men without classic risk factors for atherosclerosis. Stroke, 32(11), 2472–2479.
Napoli, C., de Nigris, F., & Palinski, W. (2001). Multiple role of reactive oxygen species in the arterial wall. Journal of Cellular Biochemistry, 82(4), 674–682.
Palinski, W., & Napoli, C. (2002). The fetal origins of atherosclerosis: maternal hypercholesterolemia, and cholesterol-lowering or antioxidant treatment during pregnancy influence in utero programming and postnatal susceptibility to atherogenesis. FASEB Journal, 16(11), 1348–1360.
Cambonie, G., Comte, B., Yzydorczyk, C., Ntimbane, T., Germain, N., Le, N. L., et al. (2007). Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. American Journal of Physiology: Regulatory, Integrative & Comparative Physiology, 292(3), R1236–R1245.
Gluckman, P. D., Hanson, M. A., Cooper, C., & Thornburg, K. L. (2008). Effect of in utero and early-life conditions on adult health and disease. New England Journal of Medicine, 359(1), 61–73.
Tarry-Adkins, J. L., Chen, J. H., Smith, N. S., Jones, R. H., Cherif, H., & Ozanne, S. E. (2009). Poor maternal nutrition followed by accelerated postnatal growth leads to telomere shortening and increased markers of cell senescence in rat islets. FASEB Journal, 23, 1521–1528.
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H., Blauw, G. J., Susser, E. S., et al. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences of the United States of America, 105(44), 17046–17049.
Palinski, W., Miller, E., & Witztum, J. L. (1995). Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proceedings of the National Academy of Sciences of the United States of America, 92(3), 821–825.
Hansson, G. K., & Libby, P. (2006). The immune response in atherosclerosis: a double-edged sword. Nature Reviews Immunology, 6(7), 508–519.
Liguori, A., D'Armiento, F. P., Palagiano, A., Palinski, W., & Napoli, C. (2008). Maternal C-reactive protein and developmental programming of atherosclerosis. American Journal of Obstetrics & Gynecology, 198(3), 281–285.
Lindegaard, M. L., Svarrer, E. M., Damm, P., Mathiesen, E. R., & Nielsen, L. B. (2008). Increased LDL cholesterol and CRP in infants of mothers with type 1 diabetes. Diabetes/Metabolism Research & Reviews, 24(6), 465–471.
Eriksson, U. J., & Borg, L. A. (1991). Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia, 34(5), 325–331.
Yang, Z., Knight, C. A., Mamerow, M. M., Vickers, K., Penn, A., Postlethwait, E. M., et al. (2004). Prenatal environmental tobacco smoke exposure promotes adult atherogenesis and mitochondrial damage in apolipoprotein E-/- mice fed a chow diet. Circulation, 110(24), 3715–3720.
Noakes, P. S., Holt, P. G., & Prescott, S. L. (2003). Maternal smoking in pregnancy alters neonatal cytokine responses. Allergy, 58(10), 1053–1058.
Stick, S. M., Burton, P. R., Gurrin, L., Sly, P. D., & LeSouef, P. N. (1996). Effects of maternal smoking during pregnancy and a family history of asthma on respiratory function in newborn infants. Lancet, 348(9034), 1060–1064.
Sartiani, L., Cerbai, E., Lonardo, G., DePaoli, P., Tattoli, M., Cagiano, R., et al. (2004). Prenatal exposure to carbon monoxide affects postnatal cellular electrophysiological maturation of the rat heart: a potential substrate for arrhythmogenesis in infancy. Circulation, 109(3), 419–423.
Lawlor, D. A., Najman, J. M., Sterne, J., Williams, G. M., Ebrahim, S., & Davey, S. G. (2004). Associations of parental, birth, and early life characteristics with systolic blood pressure at 5 years of age: findings from the Mater-University study of pregnancy and its outcomes. Circulation, 110(16), 2417–2423.
Lindegaard, M. L., Wassif, C. A., Vaisman, B., Amar, M., Wasmuth, E. V., Shamburek, R., et al. (2008). Characterization of placental cholesterol transport: ABCA1 is a potential target for in utero therapy of Smith-Lemli-Opitz syndrome. Human Molecular Genetics, 17(23), 3806–3813.
Montoudis, A., Simoneau, L., Brissette, L., Forest, J. C., Savard, R., & Lafond, J. (1999). Impact of a cholesterol enriched diet on maternal and fetal plasma lipids and fetal deposition in pregnant rabbits. Life Sciences, 64(26), 2439–2450.
McConihay, J. A., Horn, P. S., & Woollett, L. A. (2001). Effect of maternal hypercholesterolemia on fetal sterol metabolism in the Golden Syrian hamster. Journal of Lipid Research, 42(7), 1111–1119.
Stefulj, J., Panzenboeck, U., Becker, T., Hirschmugl, B., Schweinzer, C., Lang, I., et al. (2009). Human endothelial cells of the placental barrier efficiently deliver cholesterol to the fetal circulation via ABCA1 and ABCG1. Circulation Research, 104(5), 600–608.
Palinski, W. (2009). Maternal-fetal cholesterol transport in the placenta: good, bad, and target for modulation. Circulation Research, 104(5), 569–571.
Liguori, A., D'Armiento, F. P., Palagiano, A., Balestrieri, M. L., Williams-Ignarro, S., de Nigris, F., et al. (2007). Effect of gestational hypercholesterolaemia on omental vasoreactivity, placental enzyme activity and transplacental passage of normal and oxidised fatty acids. BJOG, 114(12), 1547–1556.
Khan, I. Y., Taylor, P. D., Dekou, V., Seed, P. T., Lakasing, L., Graham, D., et al. (2003). Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension, 41(1), 168–175.
Samuelsson, A. M., Matthews, P. A., Argenton, M., Christie, M. R., McConnell, J. M., Jansen, E. H., et al. (2008). Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension, 51(2), 383–392.
Torrens, C., Brawley, L., Barker, A. C., Itoh, S., Poston, L., & Hanson, M. A. (2003). Maternal protein restriction in the rat impairs resistance artery but not conduit artery function in pregnant offspring. Journal of Physiology, 547(Pt 1), 77–84.
Bogdarina, I., Welham, S., King, P. J., Burns, S. P., & Clark, A. J. (2007). Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circulation Research, 100(4), 520–526.
Di Nicolantonio, R., Hoy, K., Spargo, S., & Morgan, T. O. (1990). Perinatal salt intake alters blood pressure and salt balance in hypertensive rats. Hypertension, 15(2), 177–182.
Contreras, R. J., Wong, D. L., Henderson, R., Curtis, K. S., & Smith, J. C. (2000). High dietary NaCl early in development enhances mean arterial pressure of adult rats. Physiology & Behavior, 71(1–2), 173–181.
National Children's Study. http://www.nationalchildrensstudy.gov/Pages/default.aspx (accessed 03/08/2009).
Trasande, L., Cronk, C., Durkin, M., Weiss, M., Schoeller, D. A., Gall, E. A., et al. (2009). Environment and obesity in the National Children's Study. Environmental Health Perspectives, 117(2), 159–166.
Landrigan, P. J., Trasande, L., Thorpe, L. E., Gwynn, C., Lioy, P. J., D'Alton, M. E., et al. (2006). The National Children's Study: a 21-year prospective study of 100,000 American children. Pediatrics, 118(5), 2173–2186.
Palinski, W., & Napoli, C. (2008). Impaired fetal growth, cardiovascular disease, and the need to move on. Circulation, 117(3), 341–343.
Acknowledgements
Supported by National Institutes of Health grant HL-089559 (W.P.), Progetto di Rilevanza Nazionale from the Italian Ministery of University and Research PRIN 2006 (C.N.) and an Ellison Medical Foundation Senior Scholar Award (W.P.).
Conflicts of Interest
E. Nicolaides is Managing Partner of a venture capital enterprise interested in prevention of developmental programming. W. Palinski is a co-inventor of several UCSD patents on immunomodulation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Palinski, W., Nicolaides, E., Liguori, A. et al. Influence of Maternal Dysmetabolic Conditions During Pregnancy on Cardiovascular Disease. J. of Cardiovasc. Trans. Res. 2, 277–285 (2009). https://doi.org/10.1007/s12265-009-9108-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-009-9108-7