
Abstract
Background
Factor XII (FXII) deficiency is an interesting condition that causes prolonged activated partial thromboplastin time without bleeding diathesis. FXII may be not important in hemostasis, but still plays roles in thrombosis and inflammation. In order to raise clinical awareness about this condition, we studied patients with severe FXII deficiency and their relatives.
Methods
Consecutive severely FXII deficient patients presenting from 1995 to 2020 were recruited from two medical centers in Taiwan. Index patients and their families were tested for FXII function, antigen and F12 gene. F12 variants were constructed into the pIRES-hrGFP vector and expressed on human embryonic kidney cells (HEK293T). FXII antigen and activity were analyzed.
Results
We found five severely FXII deficient patients, three women and two men, aged 44–71 years. FXII antigen results ranged from undetectable to 43.7%. Three different mutations were identified: c.1681C>A (p.Gly542Ser), c.1561G>A (p.Glu502Lys), and a novel mutation c.1556T>A (p.Leu500Gln). HEK293T cells expressed consistently low FXII activity with all mutations. FXII antigen expression was similar to the wild type in c.1681C>A (p.Gly542Ser), but reduced in c.1556T>A (p.Leu500Gln) and c.1561G>A (p.Glu502Lys).
Conclusions
We report five unrelated patients with severe FXII deficiency, one of whom carried a novel, cross-reacting material negative mutation c.1556T>A (p.Leu500Gln).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Factor XII (FXII) deficiency was first described by Ratnoff and Colopy [1]. It is not associated with excessive bleeding despite causing a marked prolongation of activated partial thromboplastin time (aPTT) [1, 2]. Reported prevalence of FXII deficiency among healthy blood donors was around 2% [3]. Without conspicuous presentations, most patients were identified incidentally, with isolated aPTT prolongation during routine or pre-operative examinations.
Although FXII deficiency does not cause significant bleeding diathesis, there have been reports showing that it is associated with recurrent abortions [4]. For decades FXII was considered to have “no function” in coagulation, but several animal studies later revealed otherwise [5, 6]. FXII makes an essential contribution to thrombosis [7,8,9], and inflammation [10]. Anti-factor XII treatment has achieved effectiveness in hereditary angioedema, a congenital complement disorder, in clinical trials [11, 12]. Another clinical trial explored the antithrombotic effects of anti-factor XII treatment among COVID-19 patients [13]. Hence, plenty of evidence suggests that FXII has significance in several aspects of human physiology, albeit not in hemostasis.
The F12 gene, which is 12 Kb in length and contains 14 exons [14], is located on chromosome 5q33-qter [15]. Inherited severe FXII deficiency is an autosomal recessive disorder with either homozygous or compound heterozygous mutations of F12. In addition, a well-known polymorphism of F12 (rs2545801, 46C/T), substitution of C to T at a non-coding area upstream of F12 starting codon, causing significant reduction of FXII protein translation, is more prevalent in Asian populations [16]. This polymorphism might contribute to the generally lower FXII level among Asians.
The intrinsic pathway of coagulation is initiated by FXII through contact with negatively charged surfaces that induces a conformational change in zymogen FXII [9]. A small amount of activated FXII (FXIIa) activates plasma prekallikrein into kallikrein, which in turn reciprocally activates more FXII. FXIIa also activates factor XI, which activates factor IX and then goes through the intrinsic pathway [9]. In hemostasis, this process may be redundant as factor IX can be activated by tissue factor-factor VIIa complex without factor XI or XII, while factor XI can also be activated by thrombin without FXII. This explains why FXII deficiency does not lead to clinically significant bleeding diathesis. However, FXIIa can also drive the classic complement pathway via activation of C1r [9]; in addition, kallikrein generated through the FXII activation loop, also plays a role in inflammation through the kallikrein kinin system and promoting fibrinolysis [17].
More than 60 mutations in the F12 gene have been reported (from the Human Gene Mutation Data Base). While FXII-deficient patients do not need additional management, they might still receive either unnecessary plasma transfusions or delays in necessary procedures due to unaware physicians. Furthermore, deficiency status might affect the effect of future FXII targeting therapies. Lack of awareness among physicians drove us to perform this study, in order to reveal more facts about this interesting but somehow neglected coagulation factor. Therefore, since 1995, we have collected patients with congenital severe FXII deficiency. Their clinical, laboratory, and genetic test results as well as cell expression study of their F12 gene mutations are presented in this report.
Materials and methods
Index patients and their families with FXII deficiency
From 1995 to 2020, we collected consecutive patients who were diagnosed as having severe FXII deficiency in National Taiwan University Hospital and Changhua Christian Hospital in Taiwan. Medical histories of patients and their families were reviewed with obtained informed consents. Their blood samples were collected and tested for FXII activity, antigen and F12 gene. FXII coagulant activity was measured by the one-stage method using silica as the activator (ACL TOP-500 analyzer). FXII antigen was determined by ELISA (Affinity Biologicals, Ancaster, ON, Canada). The full-length FXII gene including the promoter, 14 exons, and their junctions were amplified and sequenced with the same method published by Matsuki et al. [18]. The nucleotide number based on the National Center for Biotechnology Information (NCBI) Reference Sequence data of NG_00768.1 was used. Protein sequences of FXII in several species, including mouse, rat, human, and bovine, were obtained from the NCBI website. Sequence alignment was then performed by using the Multiple Sequence Alignment tool, ClustalW2 (https://www.ebi.ac.uk/Tools/msa/clustalw2/).
This study had been reviewed and approved by the institutional review board of investigator initiated clinical studies (No. 210130).
FXII expression
FXII cDNA was ligated into pIRES-hrGFP vector to construct the expression plasmid. FXII variants (wild type 1848 bp, c.1681G>A, c.1556T>A, and c.1561T>A) were generated, and the resulting gene fragments were ligated into the pIRES-hrGFP vector. Human embryonic kidney cells (HEK293T) were maintained in Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Paisley, USA), supplemented with 10% fetal calf serum and 2 mM glutamine in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Cells were evenly split into 10 cm dishes at a density of 2 × 106 cells/dish a day before transfection, and transient transfection of HEK 293 T cells with wild-type or mutant F12 variants were carried out using the jetPRIME® (Polyplus-transfection, Illkirch-Graffenstaden, France) according to the manufacturer’s instructions. The cells were then grown for 48 h in serum-free DMEM then cells were harvested. Cell lysate was analyzed for FXII antigen; medium was analyzed for FXII antigen by ELISA (Innovative Research, Novi, MI, USA) and FXII activity by one stage clotting assay (Actin-FSL, Dade Behring, IL, USA).
Results
From 1995 to 2020, we identified five consecutive but unrelated patients, three females and two males, with severe FXII deficiency. Their ages ranged from 44 to 71.
Clinical and laboratory studies
All patients had undergone some invasive procedures without excessive bleeding. Specifically, all patients had had tooth extractions, patient I had a liver biopsy, patient II had a laparoscopic cholecystectomy, patients III had a breast tumor excision, patient IV had an extracorporeal shockwave lithotomy, and patient V had a prostatectomy. These index patients have no relation to each other and there was no consanguinity in their families.
All patients had normal PT, prolonged aPTT, and persistently undetectable FXII function. FXII antigen was undetectable (<2.5%) in patient I, but was partially reduced in other patients, as shown in Table 1. Family members of index patients were also tested for F12 gene status, FXII activity and antigen, as shown in Table 2.
Genetic studies
From five families, three different mutations were identified (Table 2): a homozygous c.1561G–A transversion, resulting in a p.Glu502Lys substitution in exon 13 of the F12 gene was identified in patient I; a homozygous c.1681G–A transversion resulting in a p.Gly542Ser substitution, in exon 14 of the F12 gene was identified in patients II, III and IV; and a compound heterozygous mutation with c.1561G–A and a novel mutation, c.1556T–A transversion, resulting in a p.Leu500Gln substitution in exon 13 of the F12 gene was found in patient V. Four family members of patient V, listed in Table 2, carried the same normal heterozygous mutation of F12. The polymorphism status of 46 C/T (rs2545801) was also surveyed as shown in Table 2. Homozygous 46 C/C was found in patient I and a homozygous 46 T/T was found in patients II to V. Among family members of index patients, 46 C/T polymorphism status of the mutant allele is marked by underlining. For example, I-1 (father of patient I) is labelled 46 T/C, which indicates the mutant F12 is linked to 46C and the wild-type F12 has 46T.
FXII expression
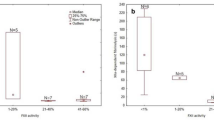
Wild-type (WT) F12 and FXII mutant variants were expressed in HEK293T cells and analyzed. The results are shown in Fig. 1. In the medium collected from cell cultures, all mutant variants expressed very low levels of FXII activity. There was no significant difference in FXII activity among the three mutations. In the medium, FXII antigen level of c.1561G>A (Glu502Lys) was lower than c1556T>A (p.Leu500Gln) which was lower than c1681C>A (p.Gly542Ser). The latter had the highest level of FXII antigen which was similar to the WT. In the lysate of cell cultures, the FXII antigen level of c.1561G>A (Glu502Lys) and c1556T>A (p.Leu500Gln) were significantly lower than WT, while c1681C>A (p.Gly542Ser) still had a similar FXII antigen level to WT.

FXII antigen expression and activity. Four different FXII variants: wild type (WT), c.1681G>A (Gly542Ser), c.1556T>A (Leu500Gln), c.1561G>A (Glu502Lys) were expressed in HEK293T. FXII antigen level in medium or cell lysate and activity in medium were measured and normalized to WT expression. The FXII antigen expression in medium and cell lysate is shown in Panel A. The FXII activity in medium is shown in panel B (WT: 100%, Gly502Lys: 10.59%, Leu500Gln: 7.44%, Glu502Lys: 3.63%)
Discussion
In this study, we reported five unrelated patients with severe FXII deficiency. One of them (patient V) had compound heterozygous mutations of F12, c.1681C>A (p.Gly542Ser) and c.1556T>A (p.Leu500Gln). The former mutation was previously reported by Zou et al. [19] from a Chinese consanguineous family. The latter mutation has not been reported before. Unlike Zou’s report [19], our patients II, III and IV, harboring the same homozygous c.1681C>A (p.Gly542Ser) mutation, did not come from consanguineous marriages and were not related to each other. This observation indicates that c.1681C>A (p.Gly542Ser) mutation could be quite common among the Taiwanese population. These three patients also had a similar FXII antigen level of around 40% which is also different from Zou’s report [19]. We are not sure about the actual cause of the difference between Zou’s and our observation. However, the tests were repeated and consistent among three patients. We are confident about our test results which were consistent with cell expression data. We found that c.1681C>A (p.Gly542Ser) expressed similar FXII antigen as WT, but patient II–IV had l FXII antigen at around 40 U/dL. We believe that this was caused by 46C/T polymorphism. While the construct of cell expression used 46C, patients II-IV all had 46 T/T linked to their mutant F12 genes. Since 46 T polymorphism has been proven to reduce FXII translation [16], it is very likely to reduce the translation with mutant F12 as well.
Patient I revealed c.1561G>A (Glu502Lys) is cross-reacting material (CRM) negative, while patients II–IV showed c.1681C>A (p.Gly542Ser) causes a CRM positive condition. Family members of patient V, who carried the novel heterozygous mutation c.1556T>A (Leu500Gln) alone, had equally low FXII antigen and activity. Hence, we concluded c.1556T>A (Leu500Gln) is also a CRM negative mutation. In patient V, with both c.1556T>A (Leu500Gln) and c.1681C>A (Gly542Ser) mutations, FXII antigen might come from the allele of c.1681C>A (Gly542Ser) alone. That would also explain why the FXII antigen of patient V was about half the level of those of patients II–IV. Findings from our patients were compatible with the cell expression result. All mutant variants expressed consistently low FXII activity. In the medium and the lysate, FXII antigen of c.1681C>A (p.Gly542Ser), the CRM positive mutation, was similar to the WT, while c.1556T>A (p.Leu500Gln) and c.1561 G>A (Glu502Lys) had significantly lower FXII antigen expression than the WT and c.1681C>A (p.Gly542Ser).
However, the cell expression of three F12 mutants still expressed detectable FXII activity at 3–10% of the WT level, but we did not find any detectable FXII activity in our patients. Meanwhile, we also found detectable FXII antigen in the cell medium and lysate of c.1561G>A (Glu502Lys) and c.1556T>A (Leu500Gln).We speculate that the experimental system may not have the same protein processing and metabolism as the conditions in which hepatocytes manufacture FXII physiologically. We surmise that FXII antigen of c.1561G>A (Glu502Lys) and c.1556T>A (Leu500Gln) may be degraded, and destroy some epitopes that were supposed to be recognized by ELISA, within the cell, so we could not find as much FXII antigen in these two mutations as in the WT lysate.
The human FXII protein consists of an epidermal growth factor-like domain, the kringle domain and catalytic domains [20]. The epidermal growth factor-like and kringle domains have the ability to bind to negatively charged activating surfaces [19]. The catalytic domain is responsible for the enzymatic activity of FXII, and the catalytic triad was identified as His393, Asp442, and Ser544 [21]. The catalytic domain of FXII shares sequence homology with other serine proteases including trypsin, elastase, chymotrypsin, and tissue-type plasminogen activator [22]. However, no three-dimensional structure has yet been reported for the catalytic domain of FXII.
To investigate the potential role of the Leu500, Glu502, and Gly542 residues, which are all located in the catalytic domain, we collected the alignment data of the human FXII protein sequences with other species’ FXII (Fig. 2) or other serine proteases [22]. We found that the Leu500, Glu502, and Gly542 amino acids of FXII are highly conserved across several species (Fig. 2). However, Leu500 and Gly542 were conserved in trypsin-like proteins, but Glu502 was not [22]. The p.Glu502Lys [23], and p.Gly542Ser FXII gene [18] mutations have been reported previously.

Protein sequences of FXII and sequence alignment by using the multiple sequence alignment tool, ClustalW2, in several species, including bovine, humans, mouse and rat. The Leu500, Glu502 and Gly542 amino acids of FXII are highly conserved
The residue Leu500 of human FXII is identical to the bovine trypsin Gly155, which forms a hydrogen bond with Asp71 [22, 24]. These studies suggested that the Leu500Gln mutation may change from a hydrophobic Leu500 to a hydrophilic polar Gln500 with an uncharged side chain and thus may interrupt the conformation of the catalytic domain of FXII causing an unstable structure of the catalytic triad His393-Asp422–Ser544, resulting in misfolding, rapid degradation, or impaired secretion leading to very low FXII antigen as we saw in patient V.
Conclusion
We conducted a comprehensive study of five unrelated patients with severe FXII deficiency and their blood relatives. Two reported mutations c.1681C>A (Gly542Ser), c.1561G>A (Glu502Lys), and a novel mutation c.1556T>A (Leu500Gln) were identified. The novel mutation is a CRM negative (type 1) mutation.
References
Ratnoff OD, Colopy JE. A familial hemorrhagic trait associated with a deficiency of a clot-promoting fraction of plasma. J Clin Invest. 1955;34(4):602–13. https://doi.org/10.1172/JCI103109.
Schloesser M, Zeerleder S, Lutze G, Halbmayer WM, Hofferbert S, Hinney B, et al. Mutations in the human factor XII gene. Blood. 1997;90(10):3967–77.
Halbmayer WM, Haushofer A, Schon R, Mannhalter C, Strohmer E, Baumgarten K, et al. The prevalence of moderate and severe FXII (Hageman factor) deficiency among the normal population: evaluation of the incidence of FXII deficiency among 300 healthy blood donors. Thromb Haemost. 1994;71(1):68–72.
Pauer H. Factor XII deficiency is strongly associated with primary recurrent abortions. Fertil Steril. 2003;80(3):590–4. https://doi.org/10.1016/s0015-0282(03)00788-x.
Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–81. https://doi.org/10.1084/jem.20050664.
Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203(3):513–8. https://doi.org/10.1084/jem.20052458.
Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood. 2012;120(22):4296–303. https://doi.org/10.1182/blood-2012-07-292094.
Key NS. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematol Am Soc Hematol Educ Program. 2014;2014(1):66–70. https://doi.org/10.1182/asheducation-2014.1.66.
Renne T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol. 2012;34(1):31–41. https://doi.org/10.1007/s00281-011-0288-2.
Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008;359(10):1027–36. https://doi.org/10.1056/NEJMcp0803977.
Kalinin DV (2021) Factor XII(a) inhibitors: a review of the patent literature. Expert Opin Ther Pat. https://doi.org/10.1080/13543776.2021.1945580.
A Study to Investigate CSL312 in Subjects With Hereditary Angioedema (HAE). https://ClinicalTrials.gov/show/NCT03712228.
Treatment With CSL312 in Adults With Coronavirus Disease 2019 (COVID-19). https://ClinicalTrials.gov/show/NCT04409509.
Cool DE, MacGillivray RT. Characterization of the human blood coagulation factor XII gene. Intron/exon gene organization and analysis of the 5′-flanking region. J Biol Chem 1987;262(28):13662–73.
Royle NJ, Nigli M, Cool D, MacGillivray RT, Hamerton JL. Structural gene encoding human factor XII is located at 5q33-qter. Somat Cell Mol Genet. 1988;14(2):217–21.
Kanaji T, Okamura T, Osaki K, Kuroiwa M, Shimoda K, Hamasaki N, et al. A common genetic polymorphism (46 C to T substitution) in the 5′-untranslated region of the coagulation factor XII gene is associated with low translation efficiency and decrease in plasma factor XII level. Blood. 1998;91(6):2010–4.
Bjorkqvist J, Nickel KF, Stavrou E, Renne T. In vivo activation and functions of the protease factor XII. Thromb Haemost. 2014;112(5):868–75. https://doi.org/10.1160/TH14-04-0311.
Matsuki E, Miyakawa Y, Okamoto S. A novel factor XII mutation, FXII R84P, causing factor XII deficiency in a patient with hereditary spastic paraplegia. Blood Coagul Fibrinol. 2011;22(3):227–30. https://doi.org/10.1097/MBC.0b013e328343f928.
Zou A, Wang M, Jin Y, Cheng X, Su K, Yang L. Genetic analysis of a novel missense mutation (Gly542Ser) with factor XII deficiency in a Chinese patient of consanguineous marriage. Int J Hematol. 2018;107(4):436–41. https://doi.org/10.1007/s12185-017-2393-z.
Citarella F, Ravon DM, Pascucci B, Felici A, Fantoni A, Hack CE. Structure/function analysis of human factor XII using recombinant deletion mutants. Evidence for an additional region involved in the binding to negatively charged surfaces. Eur J Biochem. 1996;238(1):240–9. https://doi.org/10.1111/j.1432-1033.1996.0240q.x.
Shan J, Baguinon M, Zheng L, Krishnamoorthi R. Expression, refolding, and activation of the catalytic domain of human blood coagulation factor XII. Protein Expr Purif. 2003;27(1):143–9. https://doi.org/10.1016/s1046-5928(02)00608-3.
Cool DE, Edgell CJ, Louie GV, Zoller MJ, Brayer GD, MacGillivray RT. Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta-factor XIIa. J Biol Chem. 1985;260(25):13666–76.
Yang L, Wang Y, Zhou J, Cheng X, Hao X, Xie H, et al. Identification of genetic defects underlying FXII deficiency in four unrelated Chinese patients. Acta Haematol. 2016;135(4):238–40. https://doi.org/10.1159/000444209.
Bartunik HD, Summers LJ, Bartsch HH. Crystal structure of bovine beta-trypsin at 1.5 A resolution in a crystal form with low molecular packing density. Active site geometry, ion pairs and solvent structure. J Mol Biol. 1989;210(4):813–28. https://doi.org/10.1016/0022-2836(89)90110-1.
Author information
Authors and Affiliations
Contributions
M.C.S. initiated, designed and supervised the whole study; S.C.C. analyzed all data and wrote the manuscript, C.Y.Y., C.H.P., S.F.K., J.S.L., P.T.L., and H.C.L. helped the experiment, C.Y.L., H.Y.L. and H.N.H. helped collection of subjects’ samples and clinical information.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Chou, SC., Lin, CY., Lin, HY. et al. Characterization of congenital factor XII deficiency in Taiwanese patients: identification of one novel and one common mutation. Int J Hematol 116, 528–533 (2022). https://doi.org/10.1007/s12185-022-03390-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-022-03390-0