
Abstract
TAL1/SCL is a prime example of an oncogenic transcription factor that is abnormally expressed in acute leukemia due to the replacement of regulator elements. This gene has also been recognized as an essential regulator of hematopoiesis. TAL1 expression is strictly regulated in a lineage- and stage-specific manner. Such precise control is crucial for the switching of the transcriptional program. The misexpression of TAL1 in immature thymocytes leads to a widespread series of orchestrated downstream events that affect several different cellular machineries, resulting in a lethal consequence, namely T-cell acute lymphoblastic leukemia (T-ALL). In this article, we will discuss the transcriptional regulatory network and downstream target genes, including protein-coding genes and non-coding RNAs, controlled by TAL1 in normal hematopoiesis and T-cell leukemogenesis.
Similar content being viewed by others


Introduction
Deregulation of transcription factor genes is a hallmark of acute leukemia [1]. A number of oncogenes and tumor suppressors have been identified from the breakpoints of chromosomal translocations, many of which are also involved in normal hematopoiesis. TAL1/SCL is one example that was originally cloned from a chromosomal translocation in T-cell acute lymphoblastic leukemia (T-ALL) [2]. This gene was soon recognized as an essential regulator of both primitive and definitive hematopoiesis [3,4,5,6,7,8,9]. Studies with animal models demonstrated that the misexpression of TAL1 leads to the disruption of T-cell development and often to T-ALL [10, 11]. Early studies demonstrated that TAL1 exerts its oncogenic properties by blocking T-cell differentiation; however, the detailed transcriptional program controlled by TAL1 in human T-ALL cells has remained unclear until recently. Accumulating evidence has shown that this factor serves as a master transcription factor that induces a unique regulatory circuit and regulates a variety of downstream targets that are orchestrated and affect several different cellular machineries required for the development and/or maintenance of T-ALL.
Overview of T-ALL
T-ALL is a hematological malignancy that arises from immature T-cell precursors (T-lymphoblasts) [12, 13]. T-ALL primarily occurs in young children but is also found in adults. The introduction of highly intensive chemotherapy has achieved a 75–90% 5-year event-free survival rate for pediatric T-ALL cases [14]. However, short-term and long-term adverse effects are still major problems associated with T-ALL therapy.
A number of chromosomal and genetic abnormalities have been identified in T-ALL, many of which affect genes encoding transcription factors [15]. In early studies, several genes were found to be overexpressed as a result of chromosomal translocations involving the T-cell receptor (TCR) regulatory element, which includes TAL1, TAL2, LYL1, LMO2, TLX1/HOX11 and TLX3/HOX11L2 [2, 16,17,18,19,20]. Subsequent studies have demonstrated that these genes act as oncogenes in T-ALL, and many are also essential regulators of normal hematopoiesis. Importantly, gene expression profiles have shown that T-ALL cases can be classified into several subgroups that are mutually exclusive, based on the expression of oncogenic transcription factors [21,22,23]: (1) the TAL-positive subgroup (expressing TAL1 or TAL2 together with LMO2 or LMO1); (2) the TLX-positive subgroup (expressing TLX1 or TLX3); (3) the NKX2-1-positive subgroup; (4) the HOXA-positive subgroup; and (5) the LMO2/LYL1-positive subgroup. These findings indicated that these transcription factors delineate distinct molecular pathways in T-ALL cells (called “type A abnormalities”) [24,25,26]. Among them, TAL-positive T-ALL is the largest subgroup, accounting for 40–60% of all cases. Additionally, early T-cell precursor (ETP) ALL/immature T-ALL has been defined based on the expression of genes that are present in normal ETP cells [27, 28]. This category overlaps with the type A abnormality and thus is not an exclusive criterion.
In addition to these abnormalities, other types of genetic alterations that are commonly observed across different subgroups of T-ALL have been reported (called “type B abnormalities”) [24,25,26]. As an example, the activating mutations of NOTCH1 and inactivating mutations of FBXW7, both of which lead to the constitutive activation of NOTCH1 protein, are found in over 50% of T-ALL cases [29, 30]. NOTCH1 primarily activates the MYC oncogene in T-ALL cells, thereby driving cell proliferation [31,32,33]. Similarly, genetic mutations or deletions in the cell cycle regulator CDKN2A/CDKN2B are frequently found in T-ALL [34]. Additionally, recent genomic studies have provided a comprehensive catalog of chromosomal and genetic abnormalities involved in T-ALL, including mutations of epigenetic regulators (e.g., PHF6, EZH2, SUZ12), the PI3K-PTEN-AKT pathway (e.g., PTEN, PIK3R1), the JAK-STAT pathway (e.g., JAK3, IL7R), ribosomal genes (e.g., RPL10), USP7, BCL11B, and NRAS genes, for example [35, 36]. It should be noted that although it is not exclusive, some of these abnormalities are more frequently found in specific subgroup(s) of T-ALL (e.g., PTEN mutations in TAL-positive cases) [35, 37, 38], suggesting a potential collaborating effect between type A and type B abnormalities. Please also refer to several recent review articles for more details regarding genetic abnormalities [24,25,26].
Molecular functions and roles of TAL1 in normal hematopoiesis
TAL1 belongs to the class II bHLH family of transcription factors that can form heterodimers with the class I bHLH proteins, called E-proteins (E2A, HEB and E2-2) [39, 40]. The TAL1-E-protein dimer subsequently forms a regulatory complex with other transcription factors, including LMO (LMO1 or LMO2), GATA (GATA1, GATA2 or GATA3) and LDB1 proteins. The RUNX1 and ETS family of transcription factors often co-occupy the same regulatory elements and coordinately regulate gene expression with the TAL1 complex [41].
In normal hematopoiesis, TAL1 is required for the specification of the blood lineage and maturation of several hematopoietic cells [39]. In the adult bone marrow, TAL1 is expressed in hematopoietic stem cells (HSCs), progenitor cells and erythro-megakaryocyte lineages. Early studies using murine models demonstrated that genetic knockout of Tal1 is embryonic lethal due to a lack of primitive erythropoiesis [3, 4]. Subsequent studies revealed that Tal1 is also required for definitive hematopoiesis, in particular for the specification of blood lineages [5,6,7,8,9]. A study using conditional knockout mice indicated that Tal1 is required for the maturation of erythrocytes and megakaryocyte lineages [42]. On the other hand, there is some contradictory evidence in terms of the ability of TAL1 to affect HSC maintenance. A study using conditional knockout mice suggested that TAL1 is not essential for the function of long-term HSCs [43], while others reported that TAL1 is required for the function of short-term HSCs and progenitor cells [44]. The lack of a phenotype in adult HSCs of the knockout mice could be partially explained by functional redundancy with the LYL1 protein [45], which is another class II bHLH protein expressed in HSCs and progenitor cells. Interestingly, a recent study has shown that the introduction of TAL1 with RUNX1, GATA2, LMO2 and ERG genes is able to reprogram murine fibroblasts into hematopoietic cells that have a multilineage potential (“iHSCs”) [46]. This study demonstrated the ability of TAL1 and its regulatory partners to act as hematopoietic reprogramming factors, which can initiate and maintain the regulatory program that determines the cell identity of HSCs.
Importantly, TAL1 and other stem cell transcription factors, including LMO2, GATA2 and LYL1, are transcriptionally silenced during normal lymphocyte development [47,48,49]. The expression of murine Tal1 is not detectable in the stages of double-negative (DN) 4 or double-positive (DP) thymocytes. Although the mechanism of gene silencing of TAL1 has not been fully elucidated yet, it could be associated with the upregulation of transcriptional repressors and/or mediated by epigenetic mechanisms. In contrast, E-proteins are functionally or transcriptionally upregulated during T-cell development [40, 49]. The silencing of TAL1 and LMO2 could result in the formation of E-protein dimers, which can regulate genes required for T-cell differentiation, such as RAG1, RAG2 and PTCRA [40, 49, 50]. Thus, the stage-specific regulation of bHLH family transcription factors is critical for normal T-cell development by switching the transcriptional program.
Transcriptional targets of TAL1 in normal hematopoietic cells
In normal HSCs and progenitor cells, TAL1 regulates several genes involved in HSC or progenitor function. One example is the receptor tyrosine kinase c-KIT, which is expressed in HSCs and is essential for definitive hematopoiesis. This gene has been identified as a direct target of the TAL1 complex [51]. The expression pattern of c-KIT during T-cell development is similar to that of TAL1, which is expressed in progenitor cells and silenced during the DN stage of thymocytes [47]. TAL1 also negatively regulates the expression of CDKN1A/p21 in HSCs, which is an inhibitor of the cell cycle [52], possibly contributing to HSC quiescence. The regulation of c-KIT and CDKN1A explains in part the role of TAL1 in normal HSCs. Additionally, a recent study has shown that TAL1 directly represses the DDIT4 gene, an inhibitor of mTORC1, in HSCs and the progenitor cells [53]. Knockdown of TAL1 was shown to repress the phosphorylation of mTOR targets, such as 4EBP1 and S6K proteins, which are required for protein synthesis during cell growth and proliferation. This finding provides insights into the link between the TAL1 and mTOR signaling pathways.
On the other hand, TAL1 can regulate a different set of genes in the erythro-megakaryocyte lineages. These genes include the α- and β-globin gene clusters and its regulator, KLF1, that are uniquely expressed and required for the function of red blood cells [54,55,56]. Similarly, the EPB42 and glycophorin A (GYPA) genes, which are abundantly expressed in erythroid cells, are regulated by TAL1 [54, 57]. UBE2H, which encodes an E2-ubiquitin conjugase during erythrocyte differentiation, is a target of TAL1 [58]. Additionally, the MEF2C gene, which encodes a transcription factor, has been reported to be directly regulated by TAL1 during megakaryocytes differentiation [59].
In addition, it is noteworthy that TAL1 regulates several other hematopoietic transcription factors in normal hematopoietic cells, including LMO2, RUNX1, GATA2, MYB and FLI1 [60,61,62,63]. These transcription factors regulate each other, thus forming a densely connected autoregulatory loop structure. This transcriptional circuitry is likely important for the maintenance of gene expression programing [64]. Together with its potential as a reprogramming factor, TAL1 is considered a master transcription factor that characterizes the cell fate and identity of normal HSCs and progenitor cells.
Oncogenic roles of TAL1 in T-cell leukemogenesis
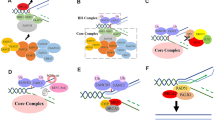
TAL1 plays an indispensable role in normal hematopoiesis, and its expression progressively decreases during T-cell development. In contrast, this factor has been implicated as an oncogene in T-ALL (Fig. 1). The TAL1 gene is overexpressed in approximately half of T-ALL cases through chromosomal translocations, intrachromosomal rearrangements, or somatic mutations in an intergenic element [2, 65,66,67,68], which in turn replaces the regulatory element.

Model of leukemic transformation induced by TAL1 in T-ALL. TAL1 is normally silenced during T-cell development. In human T-ALL cells, TAL1 is abnormally expressed due to enhancer abnormalities such as chromosomal translocation, intrachromosomal rearrangement, or mutations in the enhancer. The cells may acquire additional genetic abnormalities to be fully transformed. In TAL1-positive T-ALL cells, TAL1 protein forms a large complex with several transcription factors and coordinately regulates downstream target genes, which affect several different cellular machineries. TAL1, GATA3, RUNX1 and MYB positively regulate each other, thus forming a positive auto-regulatory loop (“core regulatory circuit”). TAL1 also inhibits E-protein functions, thereby leading to a differentiation block. Those mechanism may predispose the cells to acquire additional abnormalities such as mutations in the NOTCH1 and PIK3/AKT/PTEN pathways, which also promote cell proliferation and survival in T-ALL cells. Stages of mouse T-cell development are shown: CLP common lymphoid progenitors, DN CD4− CD8− double negative, DP, CD4+ CD8+, double positive, SP single positive (CD4+ or CD8+)
Mechanisms of activation of TAL1 in T-ALL
The chromosomal translocation involving the regulatory element of the T-cell receptor (TCR) gene [14q11.2 (TRA, TRD), 7q34 (TRB) and 7p14 (TRG)] is a hallmark of T-ALL [1]. TAL1 was originally identified as the “stem cell leukemia (SCL)” gene from the chromosomal translocation t(1;14)(p33;q11) in a T-ALL cell line [2]. This type of chromosomal translocation event leads to the abnormal expression of a translocation partner gene under the control of the TCR regulatory element. Another mechanism that drives TAL1 expression is the intrachromosomal rearrangement called “SIL-TAL1 fusion” or “SIL-TAL1 deletion” [65, 68]. In this instance, the sub-microscopic interstitial deletion including the 5′ untranslated region between the TAL1 and SIL genes leads to the abnormal expression of TAL1 under the SIL gene regulatory element. In addition to those structural abnormalities, Mansour et al. and Navarro et al. recently discovered heterozygous somatic mutations within a noncoding intergenic element prior to the TAL1 transcription start site, which introduces a MYB-binding motif and generates a new enhancer driving TAL1 expression (named MuTE) [67, 69]. This is one of the first examples of enhancer mutations in cancer. Importantly, all of these mechanisms lead to the expression of wild-type TAL1 protein. Thus, an abnormal ectopic expression but not a functional alteration is the oncogenic mechanism of TAL1.
Inhibition of E-protein functions and differentiation block
The oncogenic properties of TAL1 have also been characterized. Early studies using animal models have shown that Tal1 overexpression in murine T-cells results in a differentiation block at the DN or DP stage of thymocytes and, subsequently, T-ALL [10, 11]. One of the critical mechanisms involved is the disruption of the T-cell differentiation machinery via inhibition of E-proteins. TAL1 overexpression is thought to result in a sequestration of E-proteins into the TAL1-E-protein heterodimer and thus indirectly inhibit the transcription of genes regulated by E-protein–E-protein dimers [70]. In human and mouse T-ALL cells, the expression levels of RAG1, RAG2 and PTCRA are often repressed and can be upregulated after TAL1 knockdown [71,72,73]. Similarly, CD4, TCR-alpha and CD69 genes have been reported to be repressed by TAL1 [49, 71, 74]. Thus, ectopic TAL1 expression affects the T-cell differentiation program.
Importantly, earlier studies have shown that genetic knockout of the E-protein E2a causes a differentiation block and T-ALL [75], which are similar to the phenotypes observed in the Tal1-transgenic model. A haplo-insufficient genetic background of E-proteins (E2a or Heb) significantly accelerates tumor onset and increases overall disease penetrance in Tal1-transgenic mice [71]. Those studies clearly implicate E-proteins as tumor suppressors in the context of T-ALL. Additionally, overexpression of the mutant form of Tal1 protein, which lacks a DNA-binding ability but can still bind to E-proteins, is able to induce T-ALL [76]. Thus, this result suggests that inhibition of the E-protein-mediated transcriptional program is one of the primary mechanisms of T-cell leukemogenesis.
Collaborating oncogenic pathways of TAL1
Although these studies demonstrated the abilities of TAL1 and E-proteins to act as an oncogene and a tumor suppressor, respectively, the overexpression of TAL1 alone may not be sufficient to fully transform thymocytes. In the Tal1-transgenic mice model, only approximately 30% of animals develop T-ALL after a latent period of typically more than 100 days [71], which suggests that additional abnormalities might be required. In this regard, either the LMO1 or LMO2 gene, which encodes another component of the TAL1 complex, is abnormally expressed in almost all TAL1-positive human T-ALL cases due to enhancer abnormalities, such as mutations in the enhancer or chromosomal translocation [18, 77, 78]. In murine studies, Tal1/Lmo2-double transgenic mice show a significant acceleration of tumor onset and an increase in disease penetrance compared with single transgenic animals [11, 79, 80], demonstrating a collaborating effect. A recent structural study showed that LMO2 binding strengthens the TAL1-E2A heterodimer formation by introducing new hydrogen bonds [81]. These studies indicate that TAL1 requires either LMO1 or LMO2 to also be expressed to exert its full oncogenic ability.
Another collaborating oncogene is NOTCH1. In murine T-ALL models, the Notch1 gene is mutated in over 50% of cases, similar to human T-ALL [11, 82]. Interestingly, the frequency of Notch1 mutations in the Tal1-transgenic model is higher (over 75%) than that in T-ALL developed using other murine models (e.g., mutation of Tp53 or Atm) [82]. Several studies have demonstrated that TAL1 and NOTCH1 regulate a different set of genes [32, 83] but also share the same targets, such as CDK6 and TRIB2 [41, 84,85,86]. Thus, these two T-ALL oncogenes compensate and potentiate each other to synergistically promote T-cell leukemogenesis.
Additionally, recent genome-wide studies have revealed that several genes are more frequently mutated in the TAL1-positive T-ALL subgroup than in other subgroups [35]. For instance, mutations in the genes mediating the PI3K-PTEN-AKT-mTOR pathway, including PTEN, are observed in approximately half of TAL1-postive human T-ALL cases [35, 37, 38], while these mutations are less frequently found in TAL1-negative cases. This finding suggests potential collaborating effects or a requirement of this pathway and could also coincide with the fact that TAL1 affects the mTOR pathway in normal HSCs via the repression of DDIT4 [53]. Similarly, mutations in the USP7 gene are more frequently found in TAL1-positive cases [35], suggesting a potential collaborating role of this gene.
Transcriptional targets of TAL1 in T-ALL cells: protein-coding genes
Besides the inhibition of E-protein functions, TAL1 exerts its oncogenic ability by directly regulating target genes in T-ALL cells, including protein-coding genes and non-coding RNAs. Because TAL1 is normally silenced during T-cell development, TAL1 may not have any physiological targets in normal T-cells. Thus, activation of target genes by TAL1 in T-ALL cells could be aberrant. Such genes would be responsible for the pathogenesis of T-ALL. In this regard, we and other groups have identified several genes directly regulated by TAL1 in human T-ALL cells.
Hematopoietic transcription factors forming the core regulatory circuit
The Look, Young and Sanda laboratories and the Brand laboratory independently performed a genome-wide analysis for the identification of TAL1 targets in T-ALL cells, using ChIP-seq and gene expression analysis [41, 87]. These studies demonstrated that TAL1 regulates several other hematopoietic transcription factors, including GATA3, RUNX1, MYB and the ETS family of genes. For example, TAL1 binds to the RUNX1 enhancer (eR1), located in intron 1 of the RUNX1 gene, which has been previously reported to be active in normal HSCs and progenitors [61, 88]. Similarly, TAL1 binds to the GATA3 and MYB regulatory elements. TAL1 also binds to its own enhancer region in cases of TAL1 enhancer mutations (MuTE) [41, 67]. Independently, Thoms et al. reported that the TAL1 complex regulates the ETS family transcription factor ERG through an enhancer region in T-ALL cells [89]. ERG overexpression maintains T-ALL cell proliferation, and transgenic mice with Erg overexpression develop T-ALL. Of note, ERG directly interacts with TAL1, RUNX1, and GATA2, and this complex coordinately regulates their common targets in normal human HSCs and progenitors [63, 90]. In our previous study, other ETS family genes, including ETS1, ETV5 and ETV6, have also been found to be regulated by TAL1 in T-ALL cells [41].
Importantly, these transcription factors often co-occupy the same regulatory elements in T-ALL cells, including the enhancers of their own genes. For example, the eR1 and MuTE regions are co-bound by TAL1, GATA3, RUNX1, MYB and the ETS family proteins [41]. Similarly, GATA3 and MYB enhancers are bound by these factors. Additionally, the genes regulated after knockdown of TAL1 closely overlaps with those after knockdown of GATA3, RUNX1 or MYB [41]. Thus, these complexes of transcription factors are orchestrated in an auto-regulatory loop and coordinately regulate downstream gene expression in T-ALL cells. Of note, a recent study using murine models demonstrated that a genetic loss of Runx1 significantly delays the tumor onset of Tal1-induced T-ALL [91]. Knockdown of RUNX1 in human T-ALL cell lines inhibits cell growth. These results demonstrate the requirement of RUNX1 in the context of TAL1-positive T-ALL, which is in marked contrast to its tumor-suppressive role in myeloid malignancies.
Furthermore, it is noteworthy that the enhancers of TAL1, GATA3, RUNX1 and MYB are all associated with a super-enhancer, which is a cluster of enhancers characterized by a high level of active histone marks [92]. This finding suggests that TAL1 and its regulatory partners need to be highly expressed to maintain the transcriptional program in T-ALL cells. This type of structure has been termed as the “core regulatory circuit” and is considered to maintain the state of normal or malignant cells [41, 93, 94]. One example is Oct4, Sox2 and Nanog that form an interconnected auto-regulatory loop in embryonic stem cells [93]. These findings indicate that TAL1 functions as a master transcription factor and that the core regulatory circuit controls an extensive transcriptional regulatory network in T-ALL.
Other transcription regulators
Several other transcriptional regulators have been implicated as TAL1 targets in T-ALL cells. Our group has recently performed a comprehensive analysis using RNA-seqs after TAL1 knockdown and each of its regulatory partner proteins [95]. This analysis identified several high-confidence targets of TAL1, including ARID5B. ARID5B is a member of the ARID family of transcription factors. The enhancer controlling ARID5B expression (− 135 k enhancer) is directly activated by the TAL1 complex and is associated with a super-enhancer in T-ALL cells but not in normal T-cells. Although the molecular function of ARID5B remains unclear, its genome-wide binding profiling demonstrates that ARID5B-bound regions are predominantly associated with active histone marks in T-ALL cells. As an example, ARID5B positively regulates the expression of TAL1 and its regulatory partners (GATA3, RUNX1 and MYB) as well as the MYC oncogene in T-ALL cells. Therefore, ARID5B may serve as a transcriptional activator that enhances or consolidates the regulatory network initiated by the TAL1 complex. In addition, ARID5B overexpression in zebrafish leads to thymus retention, radio-resistance and differentiation arrest, some of which can result in T-ALL [95]. These results indicate that ARID5B plays critical oncogenic roles as a second-tier factor of TAL1 in T-ALL.
Similarly, the NKX3-1 gene, which encodes an NKL homeobox transcription factor, has been reported as a downstream target of the TAL1 complex in T-ALL cells [94,95,, 96, 97]. NKX3-1 is normally expressed in prostate and testis and plays substantial roles in several fundamental cellular processes, such as stem cell maintenance, embryogenesis and cell proliferation [98, 99]. NKX3-1 is required for T-ALL cell proliferation and maintenance in mice [96]. The downstream analysis of NKX3-1 revealed that it regulates MYCN and a homeobox gene, SIX6. These results indicate there is a critical role of NKX3-1 as a downstream target of TAL1.
The oncogene MYCN has also been reported as a TAL1 target in T-ALL cells [100]. Silencing of MYCN induces apoptosis in TAL1-positive T-ALL cells, implying its oncogenic role. MYCN is normally expressed in HSCs and progenitor cells but is silenced during the DN stage of thymocytes [47], which is similar to the TAL1 expression pattern. Thus, MYCN could be a physiological target of TAL1 in normal hematopoietic cells. However, MYCN is also overexpressed in TAL1-negative, LYL1-positive T-ALL cases that typically show the ETP/immature T-ALL phenotype [21], suggesting that MYCN could be regulated by other transcription factors in progenitor cells. In most T-ALL cases, another MYC oncogene (MYC/c-MYC) is highly expressed due to activating mutations of NOTCH1 and serves as a driver oncogene [31,32,33]. Hence, the significance of MYCN overexpression in the pathogenesis of TAL1-positive T-ALL remains unclear.
Additionally, NF-κB signaling has been reported to be regulated by the TAL1 complex in T-ALL. Poglio et al. reported that the NF-kB pathway contributes to rapid T-ALL growth in xenografts [101]. Increased NF-kB activity is observed in pre-malignant thymocytes and tumors in Tal1-transgenic mice [102]. As a potential mechanism, TAL1 has been shown to repress the transcription of the NFKB1 (p105/p50) gene by directly binding to its promoter [103]. The reduced expression of p50, the processed form of p105, results in the formation of the atypical p65/c-REL complex rather than the classic p65/p50 NF-kB dimer and thus, upregulates p65/c-REL target genes.
Cell cycle regulators
One of the common phenotypes observed in T-ALL is the abnormal proliferation of immature thymocytes. Previous studies have shown that CDK6 and its substrate Cyclin D3 are required for T-ALL development [104,105,106]. Choi et al. reported that genetic knockout of Cyclin D3 inhibits both the initiation and maintenance of murine T-ALL driven by Notch1 [106]. More recently, the same group has reported that most T-ALL cells predominantly express CDK6 and Cyclin D3, while a low level of other D-type cyclins and CDK4 have been found [107]. In addition to cell cycle regulation, increased expression of the Cyclin D3-CDK6 complex is associated with the capacity of enzymes involved in glycolysis, which reprograms the cell for the pentose phosphate and serine pathways rather than glycolysis.
In this regard, TAL1 has been known to directly regulate CDK6 to supplement this factor, where it can also suppress the transcription of CDKN2A/p16, which inhibits CDK6-Cyclin D3 function, in T-ALL cells [87, 108]. Our previous dataset also confirmed that both the CDK6 and CCND3 gene locus are bound by the TAL1 complex [41]. In particular, the CDK6 gene is associated with a super-enhancer and is highly expressed in TAL1-positive human T-ALL cells [92]. These studies indicate that TAL1 could promote cell proliferation via the upregulation of CDK6 and CCND3. However, it should be noted that the cell cycle arrest phenotype after TAL1 knockdown in T-ALL cells is generally very subtle [41]. This effect could be in part explained by the compensatory mechanism by CDK4 and other upstream oncogenic transcription factors. For example, the CDK6 and CDK4 genes are known to be regulated by NOTCH1 in T-ALL cells [83, 84]. Nevertheless, these findings indicate an essential role of CDK6 in the initiation and maintenance of T-ALL driven by two oncogenes.
Pro-survival factors
Resisting cell death is another important hallmark of cancer [109]. Anti-apoptotic BH3 proteins, such as BCL2, BCL-XL and MCL1, are often highly expressed in T-ALL, thereby preventing apoptotic cell death [110, 111]. It is, however, noteworthy that these proteins are also expressed at specific stages in normal T-cells. For example, BCL2 is highly expressed in normal ETP cells as well as in ETP ALL, whereas BCL-XL is highly expressed in the DP stage of normal thymocytes as well as in non-ETP T-ALL [110, 112]. In our previous study, we extensively searched for the possibility of the regulation of these BH3 proteins by TAL1, but no strong evidence was discovered to confirm this hypothesis. Thus, the upregulation of those proteins may reflect the differentiation stage, where T-ALL cells arise.
On the other hand, certain TAL1 targets have known to indirectly affect apoptosis pathways. The Sicinski laboratory has shown that the depletion of Cyclin D3 or CDK6 triggers apoptotic cell death in human T-ALL cells through an increase in reactive oxygen species [106, 107]. In addition, we previously performed a loss-of-function RNA interference screen for TAL1-positive T-ALL cells [41]. Among the high-confidence targets that were directly regulated by TAL1, the TRIB2 gene was selected as a factor that is required for cell growth and survival. TRIB2 belongs to the Tribbles gene family and encodes a pseudokinase domain that is incapable of catalyzing protein phosphorylation but may act as an adaptor protein [113]. Subsequent studies by us showed that TRIB2 enhances resistance to cytotoxic drugs and upregulates the expression of the XIAP anti-apoptotic protein but not of other anti-apoptotic proteins [114]. Interestingly, TRIB2 also positively regulates the expression of TAL1, GATA3, RUNX1 and MYB and inhibits E2A, thus indicating that this factor reinforces the TAL1-induced transcriptional program as a second tier in T-ALL cells. Of note, TRIB2 is also a NOTCH1 target in T-ALL [85, 86]. Ablation of TRIB2 shortened T-ALL tumor onset and accelerated the penetrance in a Notch-driven T-ALL mice model, possibly through the negative regulation of MAPK signaling or C/EBP-α [115, 116].
DNA damage repair factors
Mouse studies have shown that TAL1 induces T-ALL after a long latent period [71], which suggests that pre-leukemic cells initiated by TAL1 overexpression may require an accumulation of genetic abnormalities for full leukemic transformation. In general, an enhanced genotoxicity or compromised DNA repair machinery contributes to this mechanism [109]. For example, KU70/KU80 (XRCC6/XRCC5)-deficient mice develop T-cell malignancies [117], because impaired nonhomologous end joining (NHEJ) machinery facilitates chromosomal deletions, transversions and translocation in early leukemic cells. Similarly, mice deficient in Tp53 or Atm frequently develop T-cell malignancies [82, 118].
In this regard, the TOX gene has been reported as a TAL1 target in T-ALL [41, 119]. TOX protein binds directly to the dimeric KU70/80 and suppresses the recruitment of this complex to the DNA breaks. This suppression impedes the formation of the full DNA-PK complex, which is an essential machinery during the NHEJ repair. Thus, TOX upregulation possibly elevates genomic instability and expands the initiating pool of transformed clones via suppressing the NHEJ repair pathway. In a zebrafish model, overexpression of human TOX was found to accelerate Myc-driven T-ALL onset [119]. TOX sustains leukemic cell proliferation following transformation, as interpreted from its indispensable roles in T-ALL xenograft growth in mice. These studies indicated an oncogenic role of TOX in T-ALL development.
Other TAL1 target genes
Despite the regulatory roles of the TAL1 complex as well as its landmark targets having been well elucidated in T-ALL, the functions of several TAL1 downstream genes remain uncharacterized. TSPAN7/CD231 is one of the first examples reported as a TAL1 target in human T-ALL cells [120]. This gene encodes a cell surface protein that was originally named “T-cell acute lymphoblastic leukemia antigen (TALLA-1)”. Exogenous transfection of TAL1, LMO and GATA3 can induce TSPAN7 protein expression in a T-ALL cell line [121]. Importantly, TSPAN7 is specifically and highly expressed in T-ALL cells compared with normal peripheral blood [122]. Although the biological function of TSPAN7 is largely unknown, this protein can be a feasible therapeutic target for T-ALL.
Another gene that was identified as a TAL1 target in human T-ALL is the ALDH1A2 gene [123]. The TAL1 complex binds within the intron of ALDH1A2 gene, initiating the transcription of the shortest variant of this gene. This variant is specific to T-ALL and is highly expressed in T-ALL cell lines and primary cases. ALDH1A2 canonically encodes a major enzyme that synthesizes retinoic acid. ALDH1A2-knockout mice show reduced production of retinoic acid and eventually develop hypoplasia or aplasia in the thymus and parathyroid glands [124], indicating that ALDH1A2 activity is indispensable for the development of these organs. Interestingly, forced expression of ALDH1A2 enhances cell proliferation and drug resistance in chronic myeloid leukemia cells [125, 126]. Moreover, genes that are highly associated with ALDH1A2 in primary T-ALL expression profiles mostly benefit cell proliferation [127]. Treatment with an ALDH1A inhibitor and a retinoic acid signaling agonist are more effective against T-ALL than B-cell lineage malignancies [127]. These results suggest potential oncogenic roles of ALDH1A2 in T-ALL pathogenesis. It is also noteworthy that ALDH activity has been implicated as a stem cell marker in both normal and cancer stem cells.
The other example of TAL1 targets in T-ALL cells is the GIMAP gene cluster [128], which consists of seven genes (GIMAP1, GIMAP2, GIMAP4, GIMAP5, GIMAP6, GIMAP7 and GIMAP8) that belong to the same gene family. Some of the GIMAP genes have been implicated in the T-cell survival-death decision [129]; however, their molecular functions have not been elucidated yet. Our group has shown that the TAL1 complex binds to the enhancer located between the GIMAP6 and GIMAP2 genes in human T-ALL cells, and this enhancer is associated with a super-enhancer [128]. This element is also bound and regulated by NOTCH1 [130]. Interestingly, knockdown of a member of the TAL1 complex or genetic knockout of the enhancer results in the concurrent downregulation of all GIMAP genes, indicating that a single enhancer can control all GIMAP genes. Notably, this enhancer can be activated in HSCs and progenitors in zebrafish. Similar to the expression pattern of Tal1, the expression of mouse Gimap genes are found in HSCs and repressed during thymocyte differentiation. Hence, GIMAP gene expression is associated with TAL1 expression. Importantly, the concomitant overexpression of GIMAP5 and GIMAP7 significantly accelerates tumor onset of T-ALL driven by Myc in zebrafish. Although further characterization of the molecular functions of GIMAPs is required, the GIMAP genes contribute to T-cell leukemogenesis as a downstream effector of TAL1 and NOTCH1.
Transcriptional targets of TAL1 in T-ALL cells: non-coding RNAs
Over the last decade, a large proportion of the human genome (> 75%) was discovered to be actively transcribed, but not all of the resulting transcripts encode proteins [131,132,133,134]. The non-coding transcription products include microRNAs (miRNAs), long non-coding RNAs (lncRNAs), small nuclear RNAs (snRNAs) and circular RNAs (circRNAs). Recent studies have demonstrated that these non-coding substances not only embrace key functions in normal development but also play critical roles in the pathogenesis of cancer [135,136,137,138].
For example, Wendel et al. have shown that miR-19 from the miR-17 ~ 92 cluster is sufficient to promote leukemogenesis in Notch1-induced T-ALL [139]. Regarding lncRNAs, Wallaerts et al. performed an expression profile in primary T-ALL cases, which was able to classify T-ALL subgroups based on their expression pattern [140]. Trimarchi et al. have reported that LUNAR1 (leukemia-induced noncoding activator RNA1) was a NOTCH1 target that plays an oncogenic role by regulating IGF1R expression [141]. Meanwhile, Wang et al. reported NALT (Notch1-associated lncRNA in T-ALL) as a lncRNA-regulating NOTCH1 expression through cis-regulation, mainly involved in cell proliferation [142]. On the other hand, we have reported several miRNAs and lncRNAs as downstream of TAL1 in T-ALL cells. However, compared with protein-coding genes, very little is known regarding these molecules.
miRNAs regulated by TAL1 in T-ALL
In the case of the TAL1-positive subgroup of T-ALL, Mansour et al. along with us previously performed a comprehensive identification of miRNAs directly regulated by TAL1 [143]. This study discovered 22 miRNAs that were significantly up or downregulated after TAL1 knockdown, and among them, five (miR-223, miR-181a, miR-29c, miR-26a and miR-620) exhibited TAL1 binding near the miRNA loci. Among the shortlisted miRNAs, miR-223 was the most promising downstream target based on its differential expression after TAL1 knockdown and its contribution to cell viability of TAL1-positive T-ALL cells. The expression pattern of miR-223 is similar to that of TAL1, which is high during the ETP stage and decreases after the DN3 stage. Importantly, miR-223 inhibits the mRNA of the ubiquitin ligase FBXW7 in a sequence-specific manner, which results in upregulation of its substrate oncoproteins including MYC, MYB, Cyclin E and NOTCH1. Hence, the TAL1-miR223 axis supports cell proliferation and survival by indirectly regulating these oncoproteins.
lncRNAs regulated by TAL1 in T-ALL
Our group has recently performed a comprehensive identification of lncRNAs expressed in TAL1-positive T-ALL cells by developing a bioinformatics pipeline and RNA-seq analysis with deep coverage [73]. We selected a list of putative lncRNAs that are directly activated by TAL1 and their regulatory complex partners. From this study, two novel lncRNAs that are aberrantly expressed in T-ALL cells but not in normal thymocytes were identified. One lncRNA is the ARID5B Inducing Enhancer Associated Long non-coding RNA (ARIEL) (originally named XLOC_005968), which is located in the ARID5B −135 k enhancer locus and is directly activated by the TAL1 complex. This transcript was specifically expressed in several TAL1-positive T-ALL cell lines and primary leukemia samples but not in normal hematopoietic cells. The other novel lncRNA (XLOC_030252) is located between the FAM49A and MYCN gene locus and is expressed in not only TAL1-subtypes but also in TLX-subtype T-ALL samples. This lncRNA is also expressed in normal hematopoietic progenitor cells and in the early stages of thymus development, suggesting that it is a physiological target of TAL1 and may play a role in normal hematopoiesis as well.
Meanwhile, we have also reported lncRNAs that are negatively regulated by TAL1, including a known lncRNA lnc-OAZ3-2:7, which is located near the RORC gene. Both lnc-OAZ3-2:7 and RORC are expressed in normal thymocytes but are downregulated in TAL1-positive T-ALL cells, suggesting that they are repressed by TAL1, possibly through inhibition of E-proteins, and may play a role in T-cell differentiation.
Conclusions and future perspectives
TAL1 is a classic example of an oncogene that was identified from a chromosomal translocation in acute leukemia. The studies on TAL1 provided several novel insights into cancer genetics, including the discovery of intrachromosomal rearrangements and mutations in the enhancer, which replace or create a regulatory element. Furthermore, recent studies have revealed TAL1 as a master transcription factor that forms a core regulatory circuit and is driven by super-enhancers. Comprehensive analysis clarified the regulatory elements and downstream targets controlled by TAL1 in T-ALL cells. All of these studies implicate TAL1 as a critical transcription factor at the top of the transcriptional hierarchy.
However, there are several questions that remain unelucidated. First, our current understanding of the regulatory network is mostly based on the function of protein-coding genes. However, our previous analysis demonstrated that approximately half of TAL1-bound regions are located in the intergenic elements, many of which do not reside near protein-coding genes. Hence, we have not yet clarified all of the targets of TAL1. Second, related to this question, the involvement of non-coding RNAs is largely unknown. Although we have identified several miRNAs and lncRNAs that are directly regulated by TAL1 in human T-ALL cells, it is difficult to elucidate their physiological functions, because the sequence of non-coding RNAs are poorly conserved among species in general, and thus, animal models may not be used. In other words, studying non-coding RNAs is critical to explaining the pathogenesis of human T-ALL, which is not observed in animal models. Third, we have not answered the most fundamental question: why does TAL1 expression cause leukemia specifically in T-cells? To address this point, more detailed analysis of the regulatory network in the context of T-cells is necessary. Lastly, although we and others have identified several downstream targets of TAL1 that contribute to leukemogenesis, we still do not have feasible therapeutic targets. More extensive investigations are necessary to develop novel therapeutics for T-ALL.
References
Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–64.
Begley CG, Aplan PD, Davey MP, Nakahara K, Tchorz K, Kurtzberg J, et al. Chromosomal translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor delta-chain diversity region and results in a previously unreported fusion transcript. Proc Natl Acad Sci USA. 1989;86:2031–5.
Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature. 1995;373:432–4.
Robb L, Lyons I, Li R, Hartley L, Kontgen F, Harvey RP, et al. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci USA. 1995;92:7075–9.
Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57.
Drake CJ, Brandt SJ, Trusk TC, Little CD. TAL1/SCL is expressed in endothelial progenitor cells/angioblasts and defines a dorsal-to-ventral gradient of vasculogenesis. Dev Biol. 1997;192:17–30.
Gering M, Rodaway AR, Gottgens B, Patient RK, Green AR. The SCL gene specifies haemangioblast development from early mesoderm. EMBO J. 1998;17:4029–45.
Mead PE, Kelley CM, Hahn PS, Piedad O, Zon LI. SCL specifies hematopoietic mesoderm in Xenopus embryos. Development. 1998;125:2611–20.
Porcher C, Liao EC, Fujiwara Y, Zon LI, Orkin SH. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development. 1999;126:4603–15.
Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J. 1996;15:5160–6.
Tremblay M, Tremblay CS, Herblot S, Aplan PD, Hebert J, Perreault C, et al. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev. 2010;24:1093–105.
Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–43.
Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8:380–90.
Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. 2015;33:2938–48.
Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol. 2005;23:6306–15.
Mellentin JD, Smith SD, Cleary ML. lyl-1, a novel gene altered by chromosomal translocation in T cell leukemia, codes for a protein with a helix-loop-helix DNA binding motif. Cell. 1989;58:77–83.
Xia Y, Brown L, Yang CY, Tsan JT, Siciliano MJ, Espinosa R 3rd, et al. TAL2, a helix-loop-helix gene activated by the (7;9)(q34;q32) translocation in human T-cell leukemia. Proc Natl Acad Sci USA. 1991;88:11416–20.
Royer-Pokora B, Loos U, Ludwig WD. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene. 1991;6:1887–93.
Kennedy MA, Gonzalez-Sarmiento R, Kees UR, Lampert F, Dear N, Boehm T, et al. HOX11, a homeobox-containing T-cell oncogene on human chromosome 10q24. Proc Natl Acad Sci USA. 1991;88:8900–4.
Bernard OA, Busson-LeConiat M, Ballerini P, Mauchauffe M, Della Valle V, Monni R, et al. A new recurrent and specific cryptic translocation, t(5;14)(q35;q32), is associated with expression of the Hox11L2 gene in T acute lymphoblastic leukemia. Leukemia. 2001;15:1495–504.
Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87.
Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood. 2005;106:274–86.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–8.
Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35:975–83.
Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16:494–507.
Van Vlierberghe P, Pieters R, Beverloo HB, Meijerink JP. Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br J Haematol. 2008;143:153–68.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, Cheng C, Su X, Rubnitz JE, Basso G, Biondi A, Pui CH, Downing JR, Campana D. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–56.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71.
O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–24.
Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–109.
Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. 2006;103:18261–6.
Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006;26:8022–31.
Okuda T, Shurtleff SA, Valentine MB, Raimondi SC, Head DR, Behm F, et al. Frequent deletion of p16INK4a/MTS1 and p15INK4b/MTS2 in pediatric acute lymphoblastic leukemia. Blood. 1995;85:2321–30.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8.
Seki M, Kimura S, Isobe T, Yoshida K, Ueno H, Nakajima-Takagi Y, et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet. 2017;49:1274–81.
Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50.
Bornschein S, Demeyer S, Stirparo R, Gielen O, Vicente C, Geerdens E, et al. Defining the molecular basis of oncogenic cooperation between TAL1 expression and Pten deletion in T-ALL using a novel pro-T-cell model system. Leukemia. 2017;32:941
Porcher C, Chagraoui H, Kristiansen MS. SCL/TAL1: a multifaceted regulator from blood development to disease. Blood. 2017;129:2051–60.
Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol. 2005;6:1079–86.
Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–21.
Schlaeger TM, Mikkola HK, Gekas C, Helgadottir HB, Orkin SH. Tie2Cre-mediated gene ablation defines the stem-cell leukemia gene (SCL/tal1)-dependent window during hematopoietic stem-cell development. Blood. 2005;105:3871–4.
Mikkola HK, Klintman J, Yang H, Hock H, Schlaeger TM, Fujiwara Y, et al. Haematopoietic stem cells retain long-term repopulating activity and multipotency in the absence of stem-cell leukaemia SCL/tal-1 gene. Nature. 2003;421:547–51.
Curtis DJ, Hall MA, Van Stekelenburg LJ, Robb L, Jane SM, Begley CG. SCL is required for normal function of short-term repopulating hematopoietic stem cells. Blood. 2004;103:3342–8.
Souroullas GP, Salmon JM, Sablitzky F, Curtis DJ, Goodell MA. Adult hematopoietic stem and progenitor cells require either Lyl1 or Scl for survival. Cell Stem Cell. 2009;4:180–6.
Batta K, Florkowska M, Kouskoff V, Lacaud G. Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep. 2014;9:1871–84.
Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, et al. Gene expression commons: an open platform for absolute gene expression profiling. PLoS One. 2012;7:e40321.
Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol. 2014;14:529–45.
Herblot S, Steff AM, Hugo P, Aplan PD, Hoang T. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain expression. Nat Immunol. 2000;1:138–44.
Kee BL, Murre C. Induction of early B cell factor (EBF) and multiple B lineage genes by the basic helix-loop-helix transcription factor E12. J Exp Med. 1998;188:699–713.
Lécuyer E, Herblot S, Saint-Denis M, Martin R, Begley CG, Porcher C, et al. The SCL complex regulates c-kit expression in hematopoietic cells through functional interaction with Sp1. Blood. 2002;100:2430–40.
Lacombe J, Herblot S, Rojas-Sutterlin S, Haman A, Barakat S, Iscove NN, et al. Scl regulates the quiescence and the long-term competence of hematopoietic stem cells. Blood. 2010;115:792–803.
Benyoucef A, Calvo J, Renou L, Arcangeli ML, van den Heuvel A, Amsellem S, et al. The SCL/TAL1 transcription factor represses the stress protein DDiT4/REDD1 in human hematopoietic stem/progenitor cells. Stem Cells. 2015;33:2268–79.
Xu Z, Huang S, Chang LS, Agulnick AD, Brandt SJ. Identification of a TAL1 target gene reveals a positive role for the LIM domain-binding protein Ldb1 in erythroid gene expression and differentiation. Mol Cell Biol. 2003;23:7585–99.
Kassouf MT, Hughes JR, Taylor S, McGowan SJ, Soneji S, Green AL, et al. Genome-wide identification of TAL1’s functional targets: insights into its mechanisms of action in primary erythroid cells. Genome Res. 2010;20:1064–83.
Lecuyer E, Hoang T. SCL: from the origin of hematopoiesis to stem cells and leukemia. Exp Hematol. 2004;32:11–24.
Lahlil R, Lécuyer E, Herblot S, Hoang T. SCL assembles a multifactorial complex that determines glycophorin A expression. Mol Cell Biol. 2004;24:1439–52.
Lausen J, Pless O, Leonard F, Kuvardina ON, Koch B, Leutz A. Targets of the Tal1 transcription factor in erythrocytes: E2 ubiquitin conjugase regulation by Tal1. J Biol Chem. 2010;285:5338–46.
Gekas C, Rhodes KE, Gereige LM, Helgadottir H, Ferrari R, Kurdistani SK, et al. Mef2C is a lineage-restricted target of Scl/Tal1 and regulates megakaryopoiesis and B-cell homeostasis. Blood. 2009;113:3461–71.
Landry J-R, Bonadies N, Kinston S, Knezevic K, Wilson NK, Oram SH, et al. Expression of the leukemia oncogene Lmo2 is controlled by an array of tissue-specific elements dispersed over 100 kb and bound by Tal1/Lmo2, Ets, and Gata factors. Blood. 2009;113:5783–92.
Nottingham WT, Jarratt A, Burgess M, Speck CL, Cheng JF, Prabhakar S, et al. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood. 2007;110:4188–97.
Wilson NK, Miranda-Saavedra D, Kinston S, Bonadies N, Foster SD, Calero-Nieto F, et al. The transcriptional program controlled by the stem cell leukemia gene Scl/Tal1 during early embryonic hematopoietic development. Blood. 2009;113:5456–65.
Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–44.
Wilkinson AC, Gottgens B. Transcriptional regulation of haematopoietic stem cells. Adv Exp Med Biol. 2013;786:187–212.
Breit TM, Mol EJ, Wolvers-Tettero IL, Ludwig WD, van Wering ER, van Dongen JJ. Site-specific deletions involving the tal-1 and sil genes are restricted to cells of the T cell receptor alpha/beta lineage: T cell receptor delta gene deletion mechanism affects multiple genes. J Exp Med. 1993;177:965–77.
Chen Q, Cheng JT, Tasi LH, Schneider N, Buchanan G, Carroll A, et al. The tal gene undergoes chromosome translocation in T cell leukemia and potentially encodes a helix-loop-helix protein. EMBO J. 1990;9:415–24.
Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–7.
Janssen JW, Ludwig WD, Sterry W, Bartram CR. SIL-TAL1 deletion in T-cell acute lymphoblastic leukemia. Leukemia. 1993;7:1204–10.
Navarro JM, Touzart A, Pradel LC, Loosveld M, Koubi M, Fenouil R, et al. Site- and allele-specific polycomb dysregulation in T-cell leukaemia. Nat Commun. 2015;6:6094.
Park ST, Sun XH. The Tal1 oncoprotein inhibits E47-mediated transcription mechanism of inhibition. J Biol Chem. 1998;273:7030–7.
O’Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell. 2004;5:587–96.
Tremblay M, Herblot S, Lecuyer E, Hoang T. Regulation of pT alpha gene expression by a dosage of E2A, HEB, and SCL. J Biol Chem. 2003;278:12680–7.
Thi Ngoc PC, Tan SH, Tan TK, Chan MM, Li Z, Yeoh AEJ, et al. Identification of novel lncRNAs regulated by the TAL1 complex in T-cell acute lymphoblastic leukemia. Leukemia. 2018. https://doi.org/10.1038/s41375-018-0110-4.
Bernard M, Delabesse E, Smit L, Millien C, Kirsch IR, Strominger JL, et al. Helix-loop-helix (E2-5, HEB, TAL1 and Id1) protein interaction with the TCRalphadelta enhancers. Int Immunol. 1998;10:1539–49.
Bain G, Engel I, Maandag ECR, Te Riele HP, Voland JR, Sharp LL, et al. E2A deficiency leads to abnormalities in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol Cell Biol. 1997;17:4782–91.
O’Neil J, Billa M, Oikemus S, Kelliher M. The DNA binding activity of TAL-1 is not required to induce leukemia/lymphoma in mice. Oncogene. 2001;20:3897.
Abraham BJ, Hnisz D, Weintraub AS, Kwiatkowski N, Li CH, Li Z, et al. Small genomic insertions form enhancers that misregulate oncogenes. Nat Commun. 2017;8:14385.
Li Z, Abraham BJ, Berezovskaya A, Farah N, Liu Y, Leon T, et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T-ALL. Leukemia. 2017;31:2057
Larson R, Lavenir I, Larson T, Baer R, Warren A, Wadman I, et al. Protein dimerization between Lmo2 (Rbtn2) and Tal1 alters thymocyte development and potentiates T cell tumorigenesis in transgenic mice. EMBO J. 1996;15:1021–7.
Draheim KM, Hermance N, Yang Y, Arous E, Calvo J, Kelliher MA. A DNA-binding mutant of TAL1 cooperates with LMO2 to cause T cell leukemia in mice. Oncogene. 2011;30:1252–60.
El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Hall-Ponsele E, Platonova O, et al. Structural basis for LMO2-driven recruitment of the SCL:E47bHLH heterodimer to hematopoietic-specific transcriptional targets. Cell Rep. 2013;4:135–47.
O’Neil J, Calvo J, McKenna K, Krishnamoorthy V, Aster JC, Bassing CH, et al. Activating Notch1 mutations in mouse models of T-ALL. Blood. 2006;107:781–5.
Sanda T, Li X, Gutierrez A, Ahn Y, Neuberg DS, O’Neil J, et al. Interconnecting molecular pathways in the pathogenesis and drug sensitivity of T-cell acute lymphoblastic leukemia. Blood. 2010;115:1735–45.
Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, Aster JC, et al. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 2009;113:1689–98.
Keeshan K, He Y, Wouters BJ, Shestova O, Xu L, Sai H, et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell. 2006;10:401–11.
Wouters BJ, Jorda MA, Keeshan K, Louwers I, Erpelinck-Verschueren CA, Tielemans D, et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood. 2007;110:3706–14.
Palii CG, Perez-Iratxeta C, Yao Z, Cao Y, Dai F, Davison J, et al. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 2011;30:494–509.
Ng CE, Yokomizo T, Yamashita N, Cirovic B, Jin H, Wen Z, et al. A Runx1 intronic enhancer marks hemogenic endothelial cells and hematopoietic stem cells. Stem Cells. 2010;28:1869–81.
Thoms JA, Birger Y, Foster S, Knezevic K, Kirschenbaum Y, Chandrakanthan V, et al. ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood. 2011;117:7079–89.
Beck D, Thoms JA, Perera D, Schütte J, Unnikrishnan A, Knezevic K, et al. Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood. 2013;122:e12–22.
Choi A, Illendula A, Pulikkan JA, Roderick JE, Tesell J, Yu J, et al. RUNX1 is required for oncogenic Myb and Myc enhancer activity in T-cell acute lymphoblastic leukemia. Blood. 2017;130:1722–33.
Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–20.
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56.
Saint-Andre V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res. 2016;26:385–96.
Leong WZ, Tan SH, Ngoc PCT, Amanda S, Yam AWY, Liau WS, et al. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017;31:2343–60.
Kusy S, Gerby B, Goardon N, Gault N, Ferri F, Gérard D, et al. NKX3. 1 is a direct TAL1 target gene that mediates proliferation of TAL1-expressing human T cell acute lymphoblastic leukemia. J Exp Med. 2010;207:2141.
Nagel S, Ehrentraut S, Tomasch J, Lienenklaus S, Schneider B, Geffers R, et al. Transcriptional activation of prostate specific homeobox gene NKX3-1 in subsets of T-cell lymphoblastic leukemia (T-ALL). PLoS One. 2012;7:e40747.
Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Young P, et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966–77.
Korkmaz KS, Korkmaz CG, Ragnhildstveit E, Kizildag S, Pretlow TG, Saatcioglu F. Full-length cDNA sequence and genomic organization of human NKX3A—alternative forms and regulation by both androgens and estrogens. Gene. 2000;260:25–36.
Astolfi A, Vendemini F, Urbini M, Melchionda F, Masetti R, Franzoni M, et al. MYCN is a novel oncogenic target in pediatric T-cell acute lymphoblastic leukemia. Oncotarget. 2014;5:120.
Poglio S, Cahu X, Uzan B, Besnard-Guerin C, Lapillonne H, Leblanc T, et al. Rapid childhood T-ALL growth in xenograft models correlates with mature phenotype and NF-κB pathway activation but not with poor prognosis. Leukemia. 2015;29:977.
O’Neil J, Ventura J-J, Cusson N, Kelliher M. NF-κB activation in premalignant mouse tal-1/scl thymocytes and tumors. Blood. 2003;102:2593–6.
Chang P-Y, Draheim K, Kelliher MA, Miyamoto S. NFKB1 is a direct target of the TAL1 oncoprotein in human T leukemia cells. Cancer Res. 2006;66:6008–13.
Hu MG, Deshpande A, Enos M, Mao D, Hinds EA, Hu GF, et al. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009;69:810–8.
Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4:451–61.
Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22:438–51.
Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature. 2017;546:426–30.
Hansson A, Manetopoulos C, Jonsson JI, Axelson H. The basic helix-loop-helix transcription factor TAL1/SCL inhibits the expression of the p16INK4A and pTalpha genes. Biochem Biophys Res Commun. 2003;312:1073–81.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014;4:1074–87.
Feng H, Stachura DL, White RM, Gutierrez A, Zhang L, Sanda T, et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell. 2010;18:353–66.
Peirs S, Matthijssens F, Goossens S, Van de Walle I, Ruggero K, De Bock CE, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014;124:3738–47.
Yokoyama T, Nakamura T. Tribbles in disease: signaling pathways important for cellular function and neoplastic transformation. Cancer Sci. 2011;102:1115–22.
Tan SH, Yam AW, Lawton LN, Wong RW, Young RA, Look AT, et al. TRIB2 reinforces the oncogenic transcriptional program controlled by the TAL1 complex in T-cell acute lymphoblastic leukemia. Leukemia. 2016;30:959–62.
Stein SJ, Mack EA, Rome KS, Pajcini KV, Ohtani T, Xu L, et al. Trib2 suppresses tumor initiation in Notch-driven T-ALL. PLoS One. 2016;11:e0155408.
Liang KL, O’Connor C, Veiga JP, McCarthy TV, Keeshan K. TRIB2 regulates normal and stress-induced thymocyte proliferation. Cell Discov. 2016;2:15050.
Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–65.
Zha S, Bassing CH, Sanda T, Brush JW, Patel H, Goff PH, et al. ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. J Exp Med. 2010;207:1369–80.
Lobbardi R, Pinder J, Martinez-Pastor B, Theodorou M, Blackburn JS, Abraham BJ, et al. TOX regulates growth, DNA repair, and genomic instability in T-cell acute lymphoblastic leukemia. Cancer Discov. 2017;7:1336–53.
Takagi S, Fujikawa K, Imai T, Fukuhara N, Fukudome K, Minegishi M, et al. Identification of a highly specific surface marker of T-cell acute lymphoblastic leukemia and neuroblastoma as a new member of the transmembrane 4 superfamily. Int J Cancer. 1995;61:706–15.
Ono Y, Fukuhara N, Yoshie O. Transcriptional activity of TAL1 in T cell acute lymphoblastic leukemia (T-ALL) requires RBTN1 or -2 and induces TALLA1, a highly specific tumor marker of T-ALL. J Biol Chem. 1997;272:4576–81.
Orentas RJ, Nordlund J, He J, Sindiri S, Mackall C, Fry TJ, et al. Bioinformatic description of immunotherapy targets for pediatric T-cell leukemia and the impact of normal gene sets used for comparison. Front Oncol. 2014;4:134.
Ono Y, Fukuhara N, Yoshie O. TAL1 and LIM-only proteins synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute lymphoblastic leukemia by acting as cofactors for GATA3. Mol Cell Biol. 1998;18:6939–50.
Vermot J, Niederreither K, Garnier JM, Chambon P, Dolle P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc Natl Acad Sci USA. 2003;100:1763–8.
Moreb JS, Ucar D, Han S, Amory JK, Goldstein AS, Ostmark B, et al. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem Biol Interact. 2012;195:52–60.
Cheung AM, Wan TS, Leung JC, Chan LY, Huang H, Kwong YL, et al. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia. 2007;21:1423–30.
Longville BA, Anderson D, Welch MD, Kees UR, Greene WK. Aberrant expression of aldehyde dehydrogenase 1A (ALDH1A) subfamily genes in acute lymphoblastic leukaemia is a common feature of T-lineage tumours. Br J Haematol. 2015;168:246–57.
Liau WS, Tan SH, Ngoc PCT, Wang CQ, Tergaonkar V, Feng H, et al. Aberrant activation of the GIMAP enhancer by oncogenic transcription factors in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31:1798–807.
Nitta T, Takahama Y. The lymphocyte guard-IANs: regulation of lymphocyte survival by IAN/GIMAP family proteins. Trends Immunol. 2007;28:58–65.
Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci USA. 2011;108:14908–13.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–63.
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253–61.
Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407.
Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12:372–9.
Wallaert A, Durinck K, Taghon T, Van Vlierberghe P, Speleman F. T-ALL and thymocytes: a message of noncoding RNAs. J Hematol Oncol. 2017;10:66.
Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, et al. Genome-wide mapping and characterization of a Notch-regulated long non-coding RNAs in acute leukemia. Cell. 2014;158:593–606.
Wang Y, Wu P, Lin R, Rong L, Xue Y, Fang Y. LncRNA NALT interaction with NOTCH1 promoted cell proliferation in pediatric T cell acute lymphoblastic leukemia. Sci Rep. 2015;5:13749.
Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013;210:1545–57.
Acknowledgements
We thank the members of Sanda laboratory for their discussions and critical reviews. This research is supported by the National Research Foundation (NRF) and the Singapore Ministry of Education (MOE) under its Research Centres of Excellence initiative. T. S. is also supported by the NRF under its Competitive Research Programme (NRF-NRFF2013-02); the National Medical Research Council, Ministry of Health, Clinician Scientists Individual Research Grant (NMRC/CIRG/1443/2016); the RNA Biology Center at CSI Singapore, NUS, from funding by the Singapore MOE’s Tier 3 Grant (MOE2014-T3-1-006); and the US National Cancer Institute (1K99CA157951).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Tan, T.K., Zhang, C. & Sanda, T. Oncogenic transcriptional program driven by TAL1 in T-cell acute lymphoblastic leukemia. Int J Hematol 109, 5–17 (2019). https://doi.org/10.1007/s12185-018-2518-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-018-2518-z