
Abstract
Myelodysplastic syndrome (MDS) without increased blasts, i.e., low-grade MDS, is the most common subtype of pediatric MDS and has characteristics different from adult form of the disease. Although histological findings of bone marrow (BM) biopsies suggest that low-grade MDS is a morphologically distinctive entity, a subset of pediatric low-grade MDS may clinically overlap with aplastic anemia (AA), such as high likelihood of hypocellular marrow and normal karyotype. In addition, children with low-grade MDS are as likely to respond to immunosuppressive therapy as those with AA, which indicates that a part of these disorders might share a common pathogenesis, that is, T cell-mediated inhibition of hematopoiesis. In contrast, a small part of children with low-grade MDS experience disease progression to advanced MDS. Given that the clinical courses of pediatric low-grade MDS are heterogeneous, assessing prognostic values of clinical, morphological, histological and cytogenetic findings is critical. Thus far, monosomy 7 and multilineage dysplasia have been suggested as prognostic factors that could predict disease progression. Treatment strategy will be optimized based on more precise prognostic factors. In the future, molecular findings may also help prognostification in children with hypoplastic BM disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic syndrome (MDS) is a term defined as a group of clonal hematopoietic stem cell disorders characterized by cytopenia, dysplasia in any one of the myeloid lineages, ineffective hematopoiesis and a varying propensity to transform into acute myeloid leukemia (AML). MDS is uncommon in children compared to the elderly, and several differences between MDS in children and adults have been identified, such as the rarity of refractory anemia with ringed sideroblasts and MDS associated with del(5q) chromosome [1]. In addition, pediatric MDS can evolve from inherited bone marrow failure syndromes (IBMFS) such as Fanconi anemia (FA).
Considering these differences, a pediatric approach to the diagnosis and management of MDS was proposed in 2003 [1] and was incorporated into the 4th edition of the World Health Organization (WHO) classification in 2008 [2]. This classification mentioned unique characteristics of pediatric MDS without increased blasts, i.e., low-grade MDS. For example, neutropenia and thrombocytopenia are more likely to be the presenting manifestation in children in contrast to adults who often present with isolated anemia [3]. Children with low-grade MDS frequently show hypocellular marrow [3]. Consequently, “refractory cytopenia of childhood (RCC)” was introduced as a provisional entity in the 2008 WHO classification [2]. It is recommended that children who meet the criteria for refractory cytopenia with multilineage dysplasia (RCMD) be considered as RCC until the prognostic significance of a multilineage dysplasia is clarified in children [2].
This review overviews the clinical, morphological and pathobiological characteristics of low-grade MDS in childhood and discusses appropriate management and treatment strategy. Current problems concerning the concept of “RCC” are also addressed.
Clinical characteristics
Pediatric MDS is an uncommon disorder accounting for 5–10 % of all hematological malignancies in childhood [1]. Of those, low-grade MDS is the most common subtype. From February 2009 to October 2013, 1000 children who were suspected of having MDS, myeloproliferative neoplasms and bone marrow (BM) failure were prospectively registered into the Japanese Society of Pediatric Hematology/Oncology database and underwent the central review of morphology and histology [4]. Of those, 339 and 24 children were diagnosed with de novo low-grade MDS and refractory anemia with excess blasts (RAEB), respectively (Fig. 1). This indicated that the annual incidence of de novo MDS in Japan was 78 cases, more than 90 % of which were classified as low-grade MDS.

Distribution of patients with myelodysplastic syndrome, myeloproliferative neoplasms and bone marrow failure in Japan. One thousand patients prospectively underwent the central review of morphology and histology from February 2009 to October 2013. This data was provided by the courtesy of Dr. Asahito Hama
Low-grade MDS is diagnosed in all age groups with median age at diagnosis ranging from 6 to 12 years from three representative reports (Table 1) [3, 5, 6]. Boys and girls are equally affected. Children usually present with symptoms related to cytopenias such as bleeding tendency, infection and pallor. Some children have no clinical symptoms and can be diagnosed by chance. Cytopenias are generally milder in low-grade MDS than aplastic anemia (AA).
A Japanese prospective registry revealed normal karyotype in 41 out of 65 children (65 %) with low-grade MDS, whereas monosomy 7 was found in only 5 (8 %) [5].
Morphology and histology
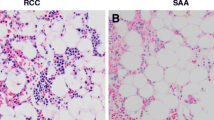
The 2008 WHO classification defines “RCC” as a childhood MDS characterized by persistent cytopenia with <5 % blasts in the BM, <2 % blasts in the peripheral blood (PB) and dysplastic changes in two or three lineages or exceeding 10 % in one single lineage [2]. BM cellularity was reduced in about half of the children with low-grade MDS [3, 5]. In children, cytopenias with decrease of BM cellularity might be caused by various underlying disorders, including AA, IBMFS and other hematological and nonhematological disorders [7]. Morphological abnormalities may also be found in these disorders. Of those, AA and IBMFS are the most common and important differential diagnoses of low-grade MDS, however, the clinical and histopathological distinction between these three disorders is challenging. Since hematopoietic cells in hypocellular low-grade MDS are often distributed in a patchy pattern (Fig. 2a), information obtained from aspiration cytology is limited [7, 8]. Therefore, BM biopsies, which mirror the topography and cellularity of the local hematopoiesis, should be performed at least twice to diagnose hypoplastic BM disorders in children [2, 7, 8]. Patchy erythropoiesis with impaired maturation, accompanied by sparsely distributed granulopoiesis, in an otherwise adipocytic BM is characteristic of hypoplastic low-grade MDS (Fig. 2b) [2, 7, 8]. Megakaryocytes are markedly decreased or absent. Immunohistochemistry is mandatory for the detection of micromegakaryocytes. A subset of children with low-grade MDS shows normo- or hypercellular BM, in which hematopoietic cells are diffusely distributed and dysplasia may be prominent (Fig. 2c, d).

Representative pictures of bone marrow (BM) biopsies obtained from patients with pediatric low-grade MDS (AS-D naphthol chloroacetate esterase stain with Giemsa staining). a, b BM specimens of hypocellular refractory cytopenia of childhood; c, d those of hypercellular refractory cytopenia with multilineage dysplasia. a Patchy distribution of erythropoiesis accompanied by sparsely distributed granulopoiesis, in an otherwise adipocytic BM. b Immature erythroid precursors form one or several islands. c Immature erythropoiesis and granulopoiesis are distributed diffusely. d Left-shifted erythroid and myeloid cells are increased. These pictures were provided by the courtesy of Dr. Masafumi Ito
Since IBMFS, such as FA and dyskeratosis congenital (DC), show overlapping morphological features with low-grade MDS and AA, IBMFS has to be excluded by careful past medical and family history, and thorough physical examination. Several laboratory studies, such as chromosome breakage test and telomere length assay, should be considered in the differential diagnosis of hypoplastic BM disorders, because children without any phenotypic features can be diagnosed with IBMFS. In fact, Yoshimi et al. [9] reported that FA and DC were found in patients with hypo- or normocellular RCC with a prevalence of 14 and 2 %, respectively.
Pathobiology
Intrinsic defects in hematopoietic stem cell caused by acquired cytogenetic and genetic abnormalities are thought as hallmark features of adult MDS. Recently, the molecular basis has been comprehensively addressed in a large cohort of MDS and AA in adults [10, 11]; however, molecular pathogenesis of pediatric MDS is still largely unknown except that spliceosome mutations are found to be rare in pediatric MDS [12].
Given that immunosuppressive therapy (IST) is effective in some children with low-grade MDS as well as MDS in adults or AA [13–15], a part of these disorders might share a common pathogenesis, that is, T cell-mediated inhibition of hematopoiesis. Indeed, T cell oligoclonality was identified in 40 % of RCC patients [16]. Minor paroxysmal nocturnal hemoglobinuria clones, which are thought to be a predictor of favorable response to IST, were also detected in 41 % of RCC patients [17]. These findings suggest that pathogenesis attributes to activated T cells in a subset of children with low-grade MDS.
Management and treatment strategy
The analysis of treatment outcome of 65 cases with low-grade MDS revealed heterogeneity in the clinical courses [5]. Approximately, a third of the patients remained stable for long periods without IST or hematopoietic stem cell transplantation (HSCT), whereas eight of 65 patients suffered from progressive disease.
Kardos et al. [3] reported that monosomy 7 significantly correlated with progression to advanced MDS. Because patients whose disease progressed before HSCT fared significantly worse, those with monosomy 7 as well as complex karyotype were encouraged to be transplanted as soon as possible. Those who suffer from transfusion dependency or severe neutropenia may also be candidates for HSCT if suitable donor is available. Because transplant-related mortality is the major cause of treatment failure in children with low-grade MDS [18], reduced-intensity conditioning (RIC) regimen is an attractive option [19, 20]; however, it remains to be established whether RIC is an appropriate regimen for children with low-grade MDS with multilineage dysplasia, hypercellularity, or unfavorable karyotype.
If children with low-grade MDS without unfavorable karyotype require therapeutic intervention but suitable donor is unavailable, IST may be an advisable option. IST consisting of antithymocyte globulin (ATG) and cyclosporine has proven to be effective in a subset of children with low-grade MDS, with response rate ranging from 30 to 70 % [5, 6, 14, 15, 21]. Some patients with chromosomal abnormalities or multilineage dysplasia were also reported to respond to IST [5, 6, 14, 15, 21]. However, patients who received IST remain at risk of clonal evolution and relapse, and the long-term failure-free survival rate was estimated as only 40–50 % [5, 21]. As reliable biomarkers that can predict response have not yet identified, candidates for IST should be selected with utmost caution.
Controversies and future directions
RCC seems a morphologically distinctive entity, but its clinical relevance has been challenged. Forester et al. [22] performed retrospective analysis of children previously diagnosed with AA and found that histologic diagnosis of RCC did not predict IST failure or clonal evolution. Hama et al. also argued that morphological classification (AA, RCC and RCMD) did not correlate with IST response or clonal evolution in the retrospective analysis. Although some clinical features of RCC may overlap with AA, these results should be interpreted with cautions, since they retrospectively analyzed a cohort of patients who had been diagnosed with AA. Another controversial point in the concept of RCC is significance of multilineage dysplasia, which correlated with deterioration of cytopenia during the watchful observation period and disease progression in our analysis [5]. To select patients who would fail IST, experience clonal evolution and require HSCT at the early phase of the disease, comprehensive prospective study assessing prognostic values of clinical, morphological, histological and cytogenetic findings and predefined treatment assignment will be needed. In the future, molecular findings may also help prognostification in children with hypoplastic BM disorders [23].
References
Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277–82.
Baumann I, Niemeyer CM, Bennett JM. Childhood myelodysplastic syndrome. In: Swerdlow SH, et al., editors. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press; 2008. p. 104–7.
Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KR, et al. Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7. Blood. 2003;102:1997–2003.
Hama A, Manabe A, Hasegawa D, Nozawa K, Okuno Y, Irie M, et al. Central morphology review of childhood bone marrow failure in Japan. Blood. 2014;124:1604 [abstract].
Hasegawa D, Chen X, Hirabayashi S, Ishida Y, Watanabe S, Zaike Y, et al. Clinical characteristics and treatment outcome in 65 cases with refractory cytopenia of childhood defined according to the WHO 2008 classification. Br J Haematol. 2014;166:758–66.
Hama A, Takahashi Y, Muramatsu H, Ito M, Narita A, Kosaka Y, et al. Comparison of long-term outcomes between children with aplastic anemia and refractory cytopenia of childhood who received immunosuppressive therapy with antithymocyte globulin and cyclosporine. Haematologica. 2015;100:1426–33.
Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematol Am Soc Hematol Educ Progr. 2011;2011:84–9.
Baumann I, Führer M, Behrendt S, Campr V, Csomor J, Furlan I, et al. Morphological differentiation of severe aplastic anaemia from hypocellular refractory cytopenia of childhood: reproducibility of histopathological diagnostic criteria. Histopathology. 2012;61:10–7.
Yoshimi A, Niemeyer C, Baumann I, Schwarz-Furlan S, Schindler D, Ebell W, et al. High incidence of Fanconi anaemia in patients with a morphological picture consistent with refractory cytopenia of childhood. Br J Haematol. 2013;160:109–11.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47.
Hirabayashi S, Flotho C, Moetter J, Heuser M, Hasle H, Gruhn B, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood. 2012;119:e96–9.
Sloand EM, Rezvani K. The role of the immune system in myelodysplasia: implications for therapy. Semin Hematol. 2008;45:39–48.
Yoshimi A, Baumann I, Fuhrer M, Bergsträsser E, Göbel U, Sykora KW, et al. Imunosuppressive therapy with anti-thymocyte globulin and cyclosporine A in selected children with hypoplastic refractory cytopenia. Haematologica. 2007;92:397–400.
Hasegawa D, Manabe A, Yagasaki H, Ohtsuka Y, Inoue M, Kikuchi A, et al. Treatment of children with refractory anemia: the Japanese childhood MDS Study Group Trial (MDS99). Pediatr Blood Cancer. 2009;53:1011–5.
Aalbers AM, van den Heuvel-Eibrink MM, Baumann I, Beverloo HB, Driessen GJ, Dworzak M, et al. T-cell receptor Vβ skewing frequently occurs in refractory cytopenia of childhood and is associated with an expansion of effector cytotoxic T cells: a prospective study by EWOG-MDS. Blood Cancer J. 2014;4:e209.
Aalbers AM, van der Velden VH, Yoshimi A, Fischer A, Noellke P, Zwaan CM, et al. The clinical relevance of minor paroxysmal nocturnal hemoglobinuria clones in refractory cytopenia of childhood: a prospective study by EWOG-MDS. Leukemia. 2014;28:189–92.
Starý J, Locatelli F, Niemeyer CM. Stem cell transplantation for aplastic anemia and myelodysplastic syndrome. Bone Marrow Transpl. 2005;35(Suppl 1):S13–6.
Strahm B, Locatelli F, Bader P, Ehlert K, Kremens B, Zintl F, et al. Reduced intensity conditioning in unrelated donor transplantation for refractory cytopenia in childhood. Bone Marrow Transpl. 2007;40:329–33.
Inagaki J, Fukano R, Kurauchi K, Noguchi M, Tanioka S, Okamura J. Hematopoietic stem cell transplantation in children with refractory cytopenia of childhood: single-center experience using high-dose cytarabine containing myeloablative and aplastic anemia oriented reduced-intensity conditioning regimens. Biol Blood Marrow Transpl. 2015;21:565–9.
Yoshimi A, van den Heuvel-Eibrink MM, Baumann I, Schwarz S, Simonitsch-Klupp I, de Paepe P, et al. Comparison of horse and rabbit antithymocyte globulin in immunosuppressive therapy for refractory cytopenia of childhood. Haematologica. 2014;99:656–63.
Forester CM, Sartain SE, Guo D, Harris MH, Weinberg OK, Fleming MD, et al. Pediatric aplastic anemia and refractory cytopenia: a retrospective analysis assessing outcomes and histomorphologic predictors. Am J Hematol. 2015;90:320–6.
Patel B, Hirsch C, Clemente M, Sekeres M, Makishima H, Maciejewski JP. Genetic and molecular characterization of myelodysplastic syndromes and related myeloid neoplasms. Int J Hematol. 2015;101:213–8.
Acknowledgments
The author would like to thank Ms. Y Imanishi for preparing and refining the patients’ data, and Drs. S Kojima, A Hama, K Nozawa and M Ito for central review of specimens. The author also thanks Dr. A Manabe for valuable advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
About this article

Cite this article
Hasegawa, D. The current perspective of low-grade myelodysplastic syndrome in children. Int J Hematol 103, 360–364 (2016). https://doi.org/10.1007/s12185-016-1965-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-1965-7