
Abstract
Interferon regulatory factor-8 (IRF8) is a transcription factor expressed in hematopoietic cells, particularly in mononuclear phagocytes [monocytes/macrophages and dendritic cells (DCs)] and their progenitors. Various studies have demonstrated that IRF8 is essential for the development of monocytes, DCs, eosinophils, and basophils. Conversely, IRF8 suppresses the generation of neutrophils. Accordingly, Irf8 −/− mice develop immunodeficiency and a chronic myeloid leukemia (CML)-like disease. Mutations and loss of expression of the human IRF8 gene are also associated with immunodeficiency and CML, respectively. Recent findings have begun to reveal the transcription factor network and epigenetic changes governed by IRF8. For example, in mononuclear phagocyte progenitors, IRF8 cooperates with PU.1 to promote the formation of promoter-distal enhancers to induce monocyte-related genes including the critical downstream transcription factor gene Klf4. On the other hand, IRF8 blocks C/EBPα activity to suppress the neutrophil differentiation program. Indeed, Irf8 −/− mononuclear phagocyte progenitors fail to efficiently generate monocytes and DCs and, instead, aberrantly give rise to neutrophils. This article provides an overview of recent advances in our understanding of the role of IRF8 in myelopoiesis and related diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interferon regulatory factor-8 (IRF8), which was originally designated interferon consensus sequence-binding protein (ICSBP), was cloned in 1990 by Ozato and colleagues as an interferon-γ (IFNγ)-inducible nuclear protein that binds to the IFN response motif in the major histocompatibility complex (MHC) class I genes [1]. The structures of the genes encoding human and mouse IRF8 are highly conserved, and their amino acid identities reach as high as 90 %.
IRF8 belongs to the IRF transcription factor family, which contains nine members in mammals [2]. IRFs have a well-conserved N-terminal DNA-binding domain (DBD) that binds to the core IRF binding motif, GAAA (Fig. 1a). Their C-terminal region carries a less well-conserved protein–protein interaction module referred to as the IRF association domain (IAD). IRF8 binds to specific DNA sequences when it forms heterodimers with partner transcription factors via its IAD and thereby acts either as an activator or a repressor [3–5] (Fig. 1b). IRF8 associates with Ets transcription factors such as PU.1 to activate transcription driven by the Ets–IRF composite element (EICE; GGAANNGAAA) or the IRF–Ets composite sequence [IECS; GAAANN(N)GGAA]. IRF8 also interacts with BATF3, although it has not yet been shown whether this occurs via the IAD, to form the IRF8–BATF3–JUN complex on the activating protein-1 (AP-1)–IRF composite element [AICE; TTTCNNNNTGA(G/C)T(C/A)A or GAAATGA(G/C)T(C/A)A] to promote gene activation [6]. In addition, IRF8 associates with other IRFs such as IRF1 and IRF2 on the IFN-stimulated response element [ISRE; (A/G)NGAAANNGAAACT]. In this case, IRF8 generally represses the transcription of ISRE-driven reporter genes.

The structure and interacting proteins of IRF8. a IRF8 interacts with C/EBPα through DBD, and with PU.1, IRF1, IRF2, and possibly BATF3 through IAD. b Transcriptional regulation by IRF8 and its interacting proteins
The phenotype of Irf8 −/− mice, generated in 1996 by Horak, Ozato, Morse, and colleagues, suggested an unpredicted role of IRF8 in myelopoiesis; Irf8 −/− mice spontaneously develop a chronic myeloid leukemia (CML)-like disease that is characterized by the systemic expansion of neutrophils followed by fatal blast crisis [7]. In addition, Irf8 −/− mice are defective in the production of interleukin-12 (IL-12) and IFNγ and are susceptible to various infections [4, 7]. Although IRF8 also regulates B cell development and innate immune responses [2, 8], this review focuses on the role of IRF8 in the development of myeloid cells and related diseases.
IRF8 expression in hematopoietic cell populations
Comprehensive analyses of IRF8 protein expression on a per cell basis in various hematopoietic cell populations [9, 10] were recently performed using IRF8–GFP chimera knock-in mice (Irf8 Irf8Gfp/WT) or intracellular immunostaining of IRF8 in mouse cells [11, 12]. IRF8 protein expression is almost absent in hematopoietic stem cells (HSCs), multipotent progenitors, and lymphoid-primed multipotent progenitors (LMPPs) (Fig. 2a). Graded expression levels of IRF8 are detected in common myeloid progenitors (CMPs) and granulocyte–monocyte progenitors (GMPs) when conventional surface markers are used. However, closer inspection has revealed that the IRF8neg fraction of CMPs and the IRF8int fraction of GMPs probably correspond to the “true” CMPs and GMPs, respectively [11, 12]. Megakaryocyte–erythroid progenitors (MEPs) are negative for IRF8 expression.

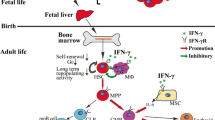
Role of IRF8 in the development of mononuclear phagocytes. a Schematic diagram of the development of monocytes and DCs in WT and Irf8 −/− mice. Irf8 −/− MDPs and cMoPs aberrantly produce large amounts of neutrophils. IRF8 protein expression levels in each population are displayed as a heat map. Mo monocyte. b Regulation of Klf4 and monocyte-related genes by IRF8. IRF8, along with PU.1, binds to the promoter-distal regions of the target genes and triggers the organization of H3K4me1 enhancers, thereby inducing Klf4 and monocyte-related genes. c Inhibition of C/EBPα activity by IRF8 in MDPs and cMoPs. IRF8 interacts with C/EBPα to inhibit the chromatin binding of C/EBPα, thereby blocking neutrophil differentiation. Thus, Irf8 −/− MDPs and cMoPs aberrantly give rise to neutrophils
High IRF8 expression is observed in the mononuclear phagocyte (i.e., monocyte/macrophage and dendritic cell [DC]) lineage (Fig. 2a). Monocyte–DC progenitors (MDPs), common monocyte progenitors (cMoPs) [13], common DC progenitors (CDPs), CD115– CDPs [plasmacytoid DC (pDC)-biased progenitors] [14], and pre-classical DCs (pre-cDCs) all highly and homogenously express IRF8. Notably, the IRF8hi subpopulations in “crude” CMPs and GMPs are likely to be mostly MDPs. Monocytes are IRF8int, and most types of macrophages (Mφs)—including Langerhans cells (LCs) and microglia—are IRF8lo/int [15]. Among DCs, pDCs, CD8α+ cDCs, and CD103+ cDCs are IRF8hi, while CD8α− (i.e., CD4+ and CD4− CD8α−) cDCs are IRF8neg/lo. Granulocyte progenitors (GPs) [16] are IRF8lo, whereas their progenies (i.e., granulocytes such as neutrophils, eosinophils, and basophils) and mast cells do not express IRF8 [17] (Fig. 3). Common lymphoid progenitors (CLPs) also express graded levels of IRF8 [12]. B cells—but not T cells unless stimulated—express IRF8. The IRF8 expression profiles largely correlate at the protein and transcript levels [18, 19].
Overall, IRF8 expression begins when progenitor cells become committed to the myeloid lineage without MEP potential and further increases when the cells progress to the mononuclear phagocyte lineage, but declines upon granulocytic differentiation.
IRF8-regulated development of monocytes and related cells
It was initially thought that Irf8 −/− mice develop immunodeficiency and CML-like disease because IRF8 regulates Mφ function and myeloid cell growth, respectively. However, a subsequent study by Scheller et al. [20] suggested not only the intrinsic leukemogenic potential and long-term reconstitution capability of Irf8 −/− progenitors, but also the role of IRF8 in the balanced generation of several myeloid cell compartments; whereas Irf8 −/− progenitors are hyperresponsive to granulocyte-Mφ colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF), their response to Mφ colony-stimulating factor (M-CSF) is significantly reduced. Moreover, Irf8 −/− colonies formed in the presence of M-CSF are mostly granulocytes. Direct evidence that IRF8 regulates hematopoietic cell differentiation was then provided by Tamura et al. who reported that reintroducing IRF8 into Irf8 −/− myeloid progenitor cell lines or freshly isolated bone marrow progenitor cells directs these cells to differentiate into functional, G0/1-arrested mature monocytes/Mφs [21, 22]. Both intact DBD and IAD of IRF8 are required to induce monocyte-related genes, such as those that encode cystatin C and cathepsins, via the IRF8–PU.1 composite elements EICE and IECS [23].
We recently found a more precise role of IRF8 in monocyte development (Fig. 2a). Irf8 −/− mice almost completely lack Ly6C+ inflammatory monocytes, and the number of Ly6C− patrolling monocytes is also reduced approximately by half in Irf8 −/− mice [5]. Close investigations of progenitor cell populations have revealed that the monocyte progenitors—i.e., MDPs and cMoPs—are not missing, but instead accumulate in Irf8 −/− mice [5, 11, 19]. In vivo cell transfer experiments show that Irf8 −/− MDPs and cMoPs are incapable of efficiently producing monocytes [11]. Thus, one of the stages where differentiation arrest occurs in Irf8 −/− mice is between cMoPs and monocytes. Notably, these data argue against the general notion that Ly6C− monocytes arise from Ly6C+ monocytes. Importantly, Gros, Cassanova, Collins, and colleagues found that the homozygous IRF8K108E mutation in humans also causes a complete lack of circulating monocytes [24].
Recent findings have also begun to reveal the transcription factor network and epigenetic changes governed by IRF8 during myeloid cell development. Chromatin immunoprecipitation with massively parallel DNA sequencing for IRF8 binding and histone modifications during IRF8-dependent in vitro monocyte differentiation have revealed that IRF8 binding occurs mostly (~80 %) at the promoter-distal regions [5]. IRF8 recognizes EICE and at a lesser frequency IECS, recruits PU.1, and promotes histone modifications characteristic of enhancers [e.g., histone H3 lysine 4 monomethylation (H3K4me1)] (Fig. 2b). Accordingly, IRF8 acts mainly as an activator during monocyte differentiation. Furthermore, Krüppel-like factor-4 (KLF4), which is essential for the development of Ly6C+ monocytes [25], was identified as a critical transcription factor that mediates the induction of “indirect” IRF8 target genes [5]. Irf8 −/− MDPs and cMoPs lack Klf4 expression, and IRF8 directly induces Klf4 probably via the IRF8-dependent distal enhancer. Introducing KLF4 into the Irf8 −/− myeloid progenitor cell line induces a subset of IRF8 target genes and thereby, partial monocyte differentiation. The KLF4-dependent IRF8 target genes are preferentially related to the basic functions of monocytes, such as inflammatory response, chemotaxis, and locomotion, while the KLF4-independent IRF8 target genes are significantly related to more advanced functions, especially antigen presentation. Indeed, IRF8 directly binds to and upregulates multiple genes that encode MHC II and the invariant chain (CD74) [5, 26]. Taken together, the evidence indicates that IRF8 cooperates with PU.1 to promote the formation of enhancers to induce various monocyte-related genes, including Klf4, and that the IRF8–KLF4 transcription factor cascade is essential for monocyte development.
Of note, growing evidence shows that tissue-resident Mφs develop from yolk sac Mφs and/or fetal definitive HSCs in embryos and are maintained in adult tissues, whereas adult HSC-derived monocytes make a negligible contribution in a steady state [27]. Nevertheless, IRF8 appears to be involved in tissue-resident Mφ development as well. The number of bone marrow resident Mφs in Irf8 −/− mice is significantly lower [28]. Even though Irf8 −/− mice display only a modest decrease in splenic red pulp Mφs, the loss of both IRF8 and structurally related IRF4 causes a significant decrease in these cells.
In a study that reported the crucial role of IRF8 in microglia activation, the expression of the microglia marker Iba1 (AIF1) was found to be diminished in Irf8 −/− spinal cords [29]. However, by using CD11b as an alternative marker, the authors have revealed that the numbers of microglia in adult Irf8 −/− mice are comparable to those in wild-type mice. Another paper demonstrated that IRF8 is required for the development of mature yolk sac Mφs (which are microglia progenitors) and suggested the reduction of Iba1+ microglia density in Irf8 −/− embryos [30]. Using Iba1 as a microglia marker in Irf8 −/− mice is debatable, because Aif1 is probably a direct target gene of IRF8 [5, 29]. At the same time, however, these findings imply the involvement of IRF8 in microgliogenesis.
Interestingly, Irf8 −/− mice harbor a high number of osteoclasts and develop osteoporosis [31]. IRF8 suppresses osteoclastogenesis by inhibiting the function and expression of NFATc1, and IRF8 expression is downregulated during receptor activator of NF-κB ligand (RANKL)-induced osteoclast differentiation. It seems reasonable to speculate that the increase in MDPs and cMoPs in Irf8 −/− bone marrow also contributes to the enhanced generation of osteoclasts.
It has been proposed that LCs should be classified as Mφs rather than DCs based on their ontogeny [32]. Although the number of LCs was initially reported to be modestly reduced in Irf8 −/− mice [33], subsequent studies on Irf8 −/− and Itgax cre Irf8 fl/fl mice [34] and humans with IRF8 mutations [24] report that IRF8 is not required to generate LCs.
Regulation of DC development by IRF8
IRF8 is also essential for the development of DCs. The numbers of pDCs and CD8α+ cDCs in lymphoid organs and of CD103+ cDCs in non-lymphoid organs are severely diminished in Irf8 −/− mice [35–39] (Fig. 2a). pDCs produce a large amount of type I IFNs upon infection, whereas CD8α+ cDCs and CD103+ cDCs induce cytotoxic T lymphocytes via cross-presentation and promote cellular immunity by producing IL-12. Accordingly, Irf8 −/− mice are immunodeficient. In humans, the homozygous IRF8K108E mutation causes the complete lack of circulating DCs and severe immunodeficiency, while the heterozygous IRF8T80A mutation is associated with the selective loss of CD11c+ CD1c+ circulating cDCs and milder immunodeficiency [24]. Both K108E and T80A mutations inhibit the DNA-binding activity of IRF8. Therefore, the overall importance of IRF8 in the development of DCs is shared between species. However, because CD11c+ CD1c+ cDCs are the human equivalent of mouse CD8α− cDCs, some species-specific differences are also conceivable.
At the progenitor level, Irf8 −/− mice accumulate MDPs but lack CDPs, CD115− CDPs, and pre-cDCs [11, 19, 40]. Thus, differentiation arrest occurs between MDPs and CDPs. Interestingly, however, CD4+ cDCs still develop in Irf8 −/− mice [23, 41]. This implies the possibility that a pathway independent of CDPs and pre-cDCs can generate CD4+ cDCs in the absence of IRF8. Alternatively, Irf8 −/− mice may harbor CDP- and pre-cDC-like cells on which surface marker expression is altered.
Recent studies have begun to uncover the transcription factor network that underpins the role of IRF8 in DC development. BXH2 mice carry a point mutation in IRF8 (R294C) that impairs its ability to form heterodimers with PU.1 on EICE [42]. In these mice, the development of CD8α+ cDCs is abolished, while the number of pDCs remains normal. Because PU.1 is required for the development of all DC subsets, one could speculate that insufficient IRF8R294C–PU.1 interaction accounts for the lack of CD8α+ cDCs in BXH2 mice. IRF8 also interacts with BATFs [6], and Batf3 −/− mice lack CD8α+ cDCs [43]. Murphy and colleagues demonstrated the importance of the IRF8–BATF interaction by showing that CD8α+ cDCs develop even in Batf3 −/− mice during infection due to the compensatory actions of other related BATFs (e.g., BATF and BATF2) [6]. Id2 is another transcription factor essential for generating CD8α+ cDCs [44]. Studies using the Irf8 −/− DC progenitor cell line and Id2–IRES–GFP reporter mice reported that IRF8 induces Id2 and then Batf3, and that BATF3 and Id2 have a synergistic effect on IRF8-directed CD8α+ cDC development [45, 46]. Importantly, it was recently reported that IRF8 regulates the formation of pDC (Flt3L-induced DC)-specific enhancers [47]. Moreover, IRF8 inhibits Cebpb expression and vice versa, thereby forming a negative feedback loop. C/EBPβ promotes the formation of monocyte-derived (GM-CSF-induced) DC-specific enhancers [47].
Notably, while IRF8 is essential for the development of CD8α+ cDCs and CD103+ cDCs, IRF4 is required for the development of CD4+ cDCs [38, 48]. The development of CD8α− CD4− (double negative, DN) cDCs requires at least one of the two IRFs [38]. While IRF8 and IRF4 share overlapping activities in the induction of genes common for all cDCs such as Ciita, each IRF possesses a distinct activity in the stimulation of subset-specific gene expression, e.g., Itgae by IRF8 and Mmp12 by IRF4, thereby leading to the generation of functionally divergent DCs [38]. Thus, the shared and specific activities of IRF8 and IRF4 govern the diversity of DCs.
As mentioned above, BXH2 mice retain pDCs. How do pDCs develop in BXH2 mice despite the fact that IRF8R294C only poorly interacts with PU.1? An intriguing possibility is that IRF8R294C retains an ability to interact with other unknown partner protein(s) to support pDC development.
Regulation of neutrophil development by IRF8
Cell lineage selection requires not only the stimulation of the gene expression program that promotes a certain cell lineage, but also suppression of the programs for other lineages. As described above, IRF8 promotes the development of monocytes and DCs. On the other hand, IRF8 inhibits neutrophil development, and Irf8 −/− mice exhibit severe neutrophilia [7, 20–22]. A human patient carrying a homozygous IRF8K108E mutation also suffers from extreme neutrophilia [24].
We recently studied the differentiation stage at which IRF8 inhibits neutrophil differentiation in mice [11]. As mentioned above, Irf8 −/− MDPs and cMoPs accumulate but fail to efficiently generate monocytes and DCs. What, then, is the fate of these progenitor cells? It has turned out that these Irf8 −/− mononuclear phagocyte progenitors aberrantly give rise to a large number of neutrophils both in vivo and in vitro (Fig. 2a). Thus, IRF8 is essential not only for bestowing DC and monocyte differentiation potential upon MDPs and cMoPs, respectively, but also for restraining these progenitors from differentiating into neutrophils. These results explain why Irf8 −/− progenitors generate neutrophils even in the presence of M-CSF [20, 22]; MDPs and cMoPs are the M-CSFR-expressing progenitor populations within Lin− cells. We infer that the production of neutrophils from MDPs and cMoPs is a major cause of neutrophilia in Irf8 −/− mice, because these progenitors are the first populations at which IRF8 is highly expressed in the myeloid lineage. It should be mentioned that all lymphoid progenitors (ALPs) from Irf8 −/− mice can also produce neutrophils [40], and the differentiation potential of Irf8 −/− GPs is biased toward neutrophils [17]. Hence, Irf8 −/− ALPs and GPs are also likely to contribute to neutrophilia.
We have also reported an underlying mechanism for IRF8-mediated inhibition of neutrophil production. IRF8 directly interacts with C/EBPα, the transcription factor known to promote neutrophil differentiation, in MDPs and cMoPs [11]. This interaction occurs through their DBDs. By doing so, IRF8 inhibits the chromatin binding and transactivation activities of C/EBPα, thereby blocking C/EBPα-induced neutrophil differentiation (Fig. 2c). Thus, Irf8 −/− MDPs and cMoPs give rise to massive numbers of neutrophils due to the derepression of C/EBPα activity (Figs. 2a, c, 3). Indeed, the partial inhibition of C/EBP activity in Irf8 −/− hematopoietic progenitors alleviates in vivo neutrophil overproduction. A more recent study from our laboratory suggests that another mechanism in GPs also exists [17], in which the transcription factor GATA2 acts downstream of IRF8 in GPs to limit their neutrophil differentiation potential (see the next section).

Role of IRF8 in the development of granulocytes and mast cells. Schematic diagram of the development of granulocytes and mast cells in WT and Irf8 −/− mice. Neutrophils are overproduced in Irf8 −/− mice from GPs and perhaps more prominently from MDPs and cMoPs. Only GMPs and GPs express intermediate levels of IRF8 among granulocyte and mast cell lineage populations
Regulation of eosinophil, basophil, and mast cell development by IRF8
Whereas IRF8 inhibits neutrophil differentiation, it promotes the generation of other types of granulocytes (e.g., eosinophils and basophils) and mast cells (Fig. 3). Irf8 −/− mice exhibit a decrease in eosinophils under physiological conditions and fail to induce eosinophilia following parasite infection despite a vigorous IL-5 response [49]. This is attributed to the reduced number and impaired differentiation potential of eosinophil lineage-committed progenitors (EoPs) in Irf8 −/− mice. Moreover, the expression of Gata1, a transcription factor gene crucial for eosinophil development, is reduced in Irf8 −/− EoPs [49].
More recently, we found that Irf8 −/− mice are basophil deficient [17]. Basophils are the least common granulocytes circulating in peripheral blood, but their critical roles in Th2-type immunity and allergic disorders have been recently clarified. Irf8 −/− mice exhibit severely reduced basophil counts. At the progenitor level, Irf8 −/− mice retain normal numbers of bone marrow GPs and splenic basophil/mast cell progenitors (BMCPs; [9]), but lack bone marrow pre-basophil and mast cell progenitor (pre-BMPs; [50]). BMCPs were originally identified as bipotential progenitors, but recent studies have shown that they give rise mostly to mast cells. Hence, the lack of basophils is explained by differentiation arrest between the GP stage and the pre-BMP stage in the bone marrow. Given that IRF8 is expressed in GPs but not their downstream populations, IRF8 appears to function in GPs. We found that Gata2, which encodes a transcription factor known to promote basophil differentiation, is induced by IRF8 in GPs and crucially acts downstream of IRF8 (Fig. 3) [17].
The complete blood count of a human patient with the homozygous IRF8K108E mutation showed a lack of basophils, but no reduction in eosinophils [24]. The reason for the latter finding is unknown, but one possibility is that IRF8 promotes eosinophil development independently of its DNA-binding activity.
IRF8’s role in mast cell development is somewhat complicated [17]. Irf8 −/− mice exhibit severe reductions in bone marrow pre-BMPs and MCPs, but retain normal numbers of splenic BMCPs, peripheral tissue MCPs, and mast cells. Nevertheless, the remaining progenitors from Irf8 −/− mice, including GPs and BMCPs, demonstrate a reduced differentiation potential toward basophils and mast cells, which is at least partially rescued by GATA2. Then, the question remains, how are Irf8 −/− peripheral tissue MCPs and mast cells generated? Although not experimentally proven, because mast cells can proliferate in peripheral tissues and are long-lived, a small number of MCPs and/or mast cells that are initially derived from Irf8 −/− BMCPs or residual pre-BMPs may proliferate and eventually reach normal levels. In addition, because IgE is known to enhance mast cell proliferation and survival, extremely high levels of IgE in Irf8 −/− mice (which can be 70 times higher than the levels in wild-type mice) may enhance the generation of peripheral tissue MCPs and mast cells, both of which express FcεRIα.
Regulation of IRF8 expression
As the critical roles of IRF8 are further revealed, our understanding of the regulatory mechanisms of IRF8 expression becomes increasingly important. Although the induction of Irf8 gene expression by IFNγ via the IFNγ-activated sequence at the promoter region has previously been described in detail [1, 51], knowledge about how IRF8 expression is controlled during myeloid cell development remains limited.
Recently, Rosenbauer and colleagues reported that a high PU.1 level directs chromatin structure remodeling at the Irf8 gene to activate Irf8 transcription in MDPs [19]. By using transgenic mouse lines harboring several copies of the phage artificial chromosome (PAC) sequence—which contains the Irf8 gene fused to the internal ribosome entry site (IRES)–YFP—these authors identified a cis-regulatory element that significantly contributes to the upregulation of Irf8 transcription in MDPs—but not in pDCs or B cells. This region is located 50-kb upstream of the Irf8 transcription start site, contains PU.1-binding sites, and requires PU.1 to activate transcription. Moreover, the −50-kb region forms a chromatin looping with the Irf8 promoter in MDPs. Wnt/β-catenin signaling was recently shown to target the Irf8 promoter and promote Irf8 expression in DCs [52]. In pDCs, TCF4/E2-2, which is a transcription factor essential for pDC development, directly induces Irf8 [53].
Several factors that repress Irf8 have been also reported, including STAT5 [54], C/EBPβ [47], GATA1 [55], WT1 [56], and BCR–ABL (see below). Interestingly, a recent study reported that myeloid-derived suppressor cells (MDSCs) develop by attenuating IRF8 expression via STAT3 and STAT5 [57].
Tumor-suppressing role of IRF8 and its relevance to CML
As already mentioned, Irf8 −/− mice develop a CML-like disease [7]. Moreover, IRF8 expression is severely reduced in CML patients and in model mice [58, 59] and the response to IFNα therapy correlates with IRF8 expression [60], suggesting the involvement of IRF8 in the pathogenesis of human CML. IRF8 inhibits myeloid cell proliferation and promotes apoptosis [20, 21, 61–64]. Moreover, Irf8 −/− progenitors possess an intrinsic leukemogenic potential and long-term reconstitution capabilities [20]. IRF8 deficiency synergizes with the oncogenes, such as AML1-ETO, NUP98-TOP1, and Meis1/3, to induce myeloblastic transformation [65–67]. Furthermore, forced IRF8 coexpression overcomes the in vitro mitogenic and anti-apoptotic activities of BCR–ABL—which is the causal oncoprotein for CML [62, 64]—and inhibits BCR–ABL-induced mouse myeloproliferative disorder in vivo [59].
While the chronic phase of CML-like disease occurs in Irf8 −/− mice very rapidly with 100 % penetrance, the transition to blast crisis occurs only after long latency at ~33 % (the original report [7]) or much lower frequency (data from our laboratory). Hence, IRF8 loss is a prerequisite, but not sufficient for malignant transformation, and requires additional genetic and/or environmental cues. Recently, cross talk between β-catenin and IRF8 was identified [68]. Preleukemic myeloproliferation in Irf8 −/− mice requires β-catenin, whereas a genetic alteration causing constitutive β-catenin activation in Irf8 −/− mice leads to the progression to blast crisis. Interestingly, activated β-catenin induces Irf8 expression [52, 68], whereas IRF8 downregulates β-catenin expression [68, 69], suggesting that IRF8 is a roadblock for β-catenin-driven leukemogenesis [68]. Importantly, BCR–ABL activates Wnt/β-catenin signaling and inhibits IRF8 expression. In fact, the disease progression in human CML coincides with the enhanced dysregulation of the two pathways.
As described above, accumulating evidence suggests that IRF8 has cell-intrinsic anti-leukemic activity and that there is an antagonistic relationship between BCR–ABL and IRF8. However, IRF8 appears to exert its anti-leukemic activity also by modulating immunity. The coexpression of IRF8 in a BCR–ABL-transformed mouse pro-B cell line induces a CD8+ cytotoxic T cell response that prevents the in vivo establishment of leukemia in a CCL6- and CCL9-dependent manner [70, 71]. Indeed, it has long been suggested that CML is highly sensitive to T cell-mediated tumor immunity. Given that IRF8 is essential for the development of DCs, another intriguing possibility is the antagonistic relationship between IRF8 and BCR–ABL in DC biology. In fact, DC counts in the peripheral blood of CML patients are significantly lower than in healthy individuals [72, 73]. Our study, which used a mouse CML model, revealed that the suppression of Irf8 by BCR–ABL is the cause of DC deficiency in CML [74]; the forced expression of IRF8 overrides BCR–ABL to rescue DC development. Notably, the effects of IRF8 supplementation are not affected by the ABL point mutation T315I that causes resistance to tyrosine kinase inhibitor (TKI) treatment. Moreover, BCR–ABL-expressing DCs rescued by IRF8 possess higher cytokine production and cytotoxic T cell induction capacity than normal DCs. Interestingly, this enhanced DC functionality is blocked by TKI treatment, suggesting that BCR–ABL harbors an immunostimulatory potential which would not be elicited during the natural course of CML or TKI therapy [74]. Thus, IRF8 appears to “convert” BCR–ABL from a DC suppressor to a DC activator. Therefore, the selective restoration of IRF8 may serve as a hallmark for new CML therapies that could overcome the problems associated with current TKI therapy, e.g., TKI resistance due to ABL mutations, CML recurrence after the discontinuation of TKI therapy, and immunosuppression by TKIs (Fig. 4).

A new strategy for CML therapy targeting the recovery of IRF8 expression. BCR–ABL inhibits DC development by suppressing IRF8 expression (right panel). Impaired DC development may cause defective immunity against leukemic cells in CML patients. By restoring IRF8 expression, the production of DCs is reversed (left panel). BCR–ABL promotes the functionality of DCs rescued by IRF8, which may contribute to eliciting effective anti-tumor immunity against CML cells
The mechanism by which BCR–ABL inhibits IRF8 gene expression then becomes an important issue. It was reported that suppressing IRF8 expression by BCR–ABL involves STAT5 and Irf8 promoter DNA methylation in primary CML cells and in BCR–ABL-positive cell lines [75, 76]. On the other hand, in another study that used BCR–ABL-transduced mouse bone marrow progenitor cells, inhibitors of either STAT5 or DNA methylation failed to rescue Irf8 expression [74]. Further investigation is required to assess whether STAT- and DNA methylation-independent mechanisms also exist.
Conclusions and perspectives
It is now clear that IRF8 regulates the development of multiple hematopoietic cell types, but then what is the common assignment for IRF8? It may be worth noting that all the cell types that require IRF8 for their development are antigen-presenting cells (APCs). DCs, monocytes, and B cells are the “professional” APCs. In addition, basophils, mast cells, and eosinophils can present antigens via MHC II when stimulated. Thus, it is tempting to speculate that a role of IRF8 common to multiple immune cell types is to generate APCs and equip these cells with antigen-presenting capabilities. Moreover, IRF8, together with the closely related IRF4, appears to confer upon DCs functional diversity, and this could be applied to other immune cell types as well.
The mechanisms of IRF8 action have also become clearer. For example, we now know that IRF8 interacts with two integral transcription factors—PU.1 and C/EBPα—to exert the dual effects on cell differentiation (e.g., stimulatory and inhibitory, respectively). However, a number of important questions remain to be addressed. Does IRF8 have lineage-specific or differentiation stage-specific modes of action to promote differentiation? What is the mechanism by which IRF8 regulates histone modifications and chromatin remodeling? What is the more comprehensive transcriptional network around IRF8? Does IRF8 connect environmental cues and the intrinsic differentiation programs? Clarifying these and other questions would contribute to our understanding of the mechanism of leukocyte development and, ultimately, the basic principles of cell differentiation.
References
Driggers PH, Ennist DL, Gleason SL, Mak WH, Marks MS, Levi BZ, et al. An interferon gamma-regulated protein that binds the interferon-inducible enhancer element of major histocompatibility complex class I genes. Proc Natl Acad Sci USA. 1990;87:3743–7.
Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–84.
Kanno Y, Levi BZ, Tamura T, Ozato K. Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J Interferon Cytokine Res. 2005;25:770–9.
Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22:145–52.
Kurotaki D, Osato N, Nishiyama A, Yamamoto M, Ban T, Sato H, et al. Essential role of the IRF8–KLF4 transcription factor cascade in murine monocyte differentiation. Blood. 2013;121:1839–49.
Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol. 2013;13:499–509.
Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–17.
Wang H, Morse HC 3rd. IRF8 regulates myeloid and B lymphoid lineage diversification. Immunol Res. 2009;43:109–17.
Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007;26:726–40.
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–61.
Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, et al. IRF8 inhibits C/EBPα activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat Commun. 2014;5:4978.
Wang H, Yan M, Sun J, Jain S, Yoshimi R, Abolfath SM, et al. A reporter mouse reveals lineage-specific and heterogeneous expression of IRF8 during lymphoid and myeloid cell differentiation. J Immunol. 2014;193:1766–77.
Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. 2013;14:821–30.
Onai N, Kurabayashi K, Hosoi-Amaike M, Toyama-Sorimachi N, Matsushima K, Inaba K, et al. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity. 2013;38:943–57.
Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–28.
Franco CB, Chen CC, Drukker M, Weissman IL, Galli SJ. Distinguishing mast cell and granulocyte differentiation at the single-cell level. Cell Stem Cell. 2010;6:361–8.
Sasaki H, Kurotaki D, Osato N, Sato H, Sasaki I, Koizumi S, et al. Transcription factor IRF8 plays a critical role in the development of murine basophils and mast cells. Blood. 2015;125:358–69.
Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol. 2012;13:888–99.
Schonheit J, Kuhl C, Gebhardt ML, Klett FF, Riemke P, Scheller M, et al. PU.1 level-directed chromatin structure remodeling at the Irf8 gene drives dendritic cell commitment. Cell Rep. 2013;3:1617–28.
Scheller M, Foerster J, Heyworth CM, Waring JF, Lohler J, Gilmore GL, et al. Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood. 1999;94:3764–71.
Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–65.
Tsujimura H, Nagamura-Inoue T, Tamura T, Ozato K. IFN consensus sequence binding protein/IFN regulatory factor-8 guides bone marrow progenitor cells toward the macrophage lineage. J Immunol. 2002;169:1261–9.
Tamura T, Thotakura P, Tanaka TS, Ko MS, Ozato K. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood. 2005;106:1938–47.
Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365:127–38.
Alder JK, Georgantas RW 3rd, Hildreth RL, Kaplan IM, Morisot S, Yu X, et al. Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. J Immunol. 2008;180:5645–52.
Marquis JF, Kapoustina O, Langlais D, Ruddy R, Dufour CR, Kim BH, et al. Interferon regulatory factor 8 regulates pathways for antigen presentation in myeloid cells and during tuberculosis. PLoS Genet. 2011;7:e1002097.
Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35.
Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, et al. Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS One. 2011;6:e25812.
Masuda T, Tsuda M, Yoshinaga R, Tozaki-Saitoh H, Ozato K, Tamura T, et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012;1:334–40.
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–80.
Zhao B, Takami M, Yamada A, Wang X, Koga T, Hu X, et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med. 2009;15:1066–71.
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14:571–8.
Schiavoni G, Mattei F, Borghi P, Sestili P, Venditti M, Morse HC 3rd, et al. ICSBP is critically involved in the normal development and trafficking of Langerhans cells and dermal dendritic cells. Blood. 2004;103:2221–8.
Chopin M, Seillet C, Chevrier S, Wu L, Wang H, Morse HC 3rd, et al. Langerhans cells are generated by two distinct PU.1-dependent transcriptional networks. J Exp Med. 2013;210:2967–80.
Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC 3rd, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8α+ dendritic cells. J Exp Med. 2002;196:1415–25.
Tsujimura H, Tamura T, Gongora C, Aliberti J, Reis e Sousa C, Sher A, et al. ICSBP/IRF-8 retrovirus transduction rescues dendritic cell development in vitro. Blood. 2003;101:961–9.
Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003;170:1131–5.
Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol. 2005;174:2573–81.
Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J Exp Med. 2010;207:823–36.
Becker AM, Michael DG, Satpathy AT, Sciammas R, Singh H, Bhattacharya D. IRF-8 extinguishes neutrophil production and promotes dendritic cell lineage commitment in both myeloid and lymphoid mouse progenitors. Blood. 2012;119:2003–12.
Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reise Sousa C, Ozato K, et al. Essential role for ICSBP in the in vivo development of murine CD8α+ dendritic cells. Blood. 2003;101:305–10.
Tailor P, Tamura T, Morse HC 3rd, Ozato K. The BXH2 mutation in IRF8 differentially impairs dendritic cell subset development in the mouse. Blood. 2008;111:1942–5.
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–100.
Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol. 2003;4:380–6.
Jackson JT, Hu Y, Liu R, Masson F, D’Amico A, Carotta S, et al. Id2 expression delineates differential checkpoints in the genetic program of CD8α+ and CD103+ dendritic cell lineages. EMBO J. 2011;30:2690–704.
Jaiswal H, Kaushik M, Sougrat R, Gupta M, Dey A, Verma R, et al. Batf3 and Id2 have a synergistic effect on Irf8-directed classical CD8α+ dendritic cell development. J Immunol. 2013;191:5993–6001.
Bornstein C, Winter D, Barnett-Itzhaki Z, David E, Kadri S, Garber M, et al. A negative feedback loop of transcription factors specifies alternative dendritic cell chromatin States. Mol Cell. 2014;56:749–62.
Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8α+ dendritic cell development. Proc Natl Acad Sci USA. 2004;101:8981–6.
Milanovic M, Terszowski G, Struck D, Liesenfeld O, Carstanjen D. IFN consensus sequence binding protein (Icsbp) is critical for eosinophil development. J Immunol. 2008;181:5045–53.
Qi X, Hong J, Chaves L, Zhuang Y, Chen Y, Wang D, et al. Antagonistic regulation by the transcription factors C/EBPα and MITF specifies basophil and mast cell fates. Immunity. 2013;39:97–110.
Kanno Y, Kozak CA, Schindler C, Driggers PH, Ennist DL, Gleason SL, et al. The genomic structure of the murine ICSBP gene reveals the presence of the gamma interferon-responsive element, to which an ISGF3α subunit (or similar) molecule binds. Mol Cell Biol. 1993;13:3951–63.
Cohen SB, Smith NL, McDougal C, Pepper M, Shah S, Yap GS, et al. β-catenin signaling drives differentiation and proinflammatory function of IRF8-dependent dendritic cells. J Immunol. 2015;194:210–22.
Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48.
Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28:509–20.
Shimokawa N, Nishiyama C, Nakano N, Maeda K, Suzuki R, Hara M, et al. Suppressive effects of transcription factor GATA-1 on cell type-specific gene expression in dendritic cells. Immunogenetics. 2010;62:421–9.
Vidovic K, Svensson E, Nilsson B, Thuresson B, Olofsson T, Lennartsson A, et al. Wilms’ tumor gene 1 protein represses the expression of the tumor suppressor interferon regulatory factor 8 in human hematopoietic progenitors and in leukemic cells. Leukemia. 2010;24:992–1000.
Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123:4464–78.
Schmidt M, Nagel S, Proba J, Thiede C, Ritter M, Waring JF, et al. Lack of interferon consensus sequence binding protein (ICSBP) transcripts in human myeloid leukemias. Blood. 1998;91:22–9.
Hao SX, Ren R. Expression of interferon consensus sequence binding protein (ICSBP) is downregulated in BCR–ABL-induced murine chronic myelogenous leukemia-like disease, and forced coexpression of ICSBP inhibits Bcr–Abl-induced myeloproliferative disorder. Mol Cell Biol. 2000;20:1149–61.
Schmidt M, Hochhaus A, Nitsche A, Hehlmann R, Neubauer A. Expression of nuclear transcription factor interferon consensus sequence binding protein in chronic myeloid leukemia correlates with pretreatment risk features and cytogenetic response to interferon-α. Blood. 2001;97:3648–50.
Gabriele L, Phung J, Fukumoto J, Segal D, Wang IM, Giannakakou P, et al. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J Exp Med. 1999;190:411–21.
Tamura T, Kong HJ, Tunyaplin C, Tsujimura H, Calame K, Ozato K. ICSBP/IRF-8 inhibits mitogenic activity of p210 Bcr/Abl in differentiating myeloid progenitor cells. Blood. 2003;102:4547–54.
Schmidt M, Bies J, Tamura T, Ozato K, Wolff L. The interferon regulatory factor ICSBP/IRF-8 in combination with PU.1 up-regulates expression of tumor suppressor p15Ink4b in murine myeloid cells. Blood. 2004;103:4142–9.
Burchert A, Cai D, Hofbauer LC, Samuelsson MK, Slater EP, Duyster J, et al. Interferon consensus sequence binding protein (ICSBP; IRF-8) antagonizes BCR/ABL and down-regulates bcl-2. Blood. 2004;103:3480–9.
Schwieger M, Lohler J, Friel J, Scheller M, Horak I, Stocking C. AML1-ETO inhibits maturation of multiple lymphohematopoietic lineages and induces myeloblast transformation in synergy with ICSBP deficiency. J Exp Med. 2002;196:1227–40.
Gurevich RM, Rosten PM, Schwieger M, Stocking C, Humphries RK. Retroviral integration site analysis identifies ICSBP as a collaborating tumor suppressor gene in NUP98-TOP1-induced leukemia. Exp Hematol. 2006;34:1192–201.
Hara T, Schwieger M, Kazama R, Okamoto S, Minehata K, Ziegler M, et al. Acceleration of chronic myeloproliferation by enforced expression of Meis1 or Meis3 in Icsbp-deficient bone marrow cells. Oncogene. 2008;27:3865–9.
Scheller M, Schonheit J, Zimmermann K, Leser U, Rosenbauer F, Leutz A. Cross talk between Wnt/β-catenin and Irf8 in leukemia progression and drug resistance. J Exp Med. 2013;210:2239–56.
Huang W, Zhou W, Saberwal G, Konieczna I, Horvath E, Katsoulidis E, et al. Interferon consensus sequence binding protein (ICSBP) decreases β-catenin activity in myeloid cells by repressing GAS2 transcription. Mol Cell Biol. 2010;30:4575–94.
Deng M, Daley GQ. Expression of interferon consensus sequence binding protein induces potent immunity against BCR/ABL-induced leukemia. Blood. 2001;97:3491–7.
Nardi V, Naveiras O, Azam M, Daley GQ. ICSBP-mediated immune protection against BCR–ABL-induced leukemia requires the CCL6 and CCL9 chemokines. Blood. 2009;113:3813–20.
Mohty M, Isnardon D, Vey N, Briere F, Blaise D, Olive D, et al. Low blood dendritic cells in chronic myeloid leukaemia patients correlates with loss of CD34+/CD38− primitive haematopoietic progenitors. Br J Haematol. 2002;119:115–8.
Boissel N, Rousselot P, Raffoux E, Cayuela JM, Maarek O, Charron D, et al. Defective blood dendritic cells in chronic myeloid leukemia correlate with high plasmatic VEGF and are not normalized by imatinib mesylate. Leukemia. 2004;18:1656–61.
Watanabe T, Hotta C, Koizumi S, Miyashita K, Nakabayashi J, Kurotaki D, et al. The transcription factor IRF8 counteracts BCR–ABL to rescue dendritic cell development in chronic myelogenous leukemia. Cancer Res. 2013;73:6642–53.
Waight JD, Banik D, Griffiths EA, Nemeth MJ, Abrams SI. Regulation of the interferon regulatory factor-8 (IRF-8) tumor suppressor gene by the signal transducer and activator of transcription 5 (STAT5) transcription factor in chronic myeloid leukemia. J Biol Chem. 2014;289:15642–52.
Hu X, Yang D, Zimmerman M, Liu F, Yang J, Kannan S, et al. IRF8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer Res. 2011;71:2882–91.
Acknowledgments
We thank Dr. Keiko Ozato for invaluable discussions and suggestions. This work was supported by Grants-in-Aids (KAKENHI) from the Japan Society for the Promotion of Science (JSPS)/Ministry of Education, Culture, Sports, Science and Technology (MEXT). S. K. is a JSPS postdoctoral fellow.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Tamura, T., Kurotaki, D. & Koizumi, Si. Regulation of myelopoiesis by the transcription factor IRF8. Int J Hematol 101, 342–351 (2015). https://doi.org/10.1007/s12185-015-1761-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1761-9