
Abstract
The OVCAR-3 cell line expressing the long (ObRb) and short (ObRt) isoforms of leptin receptor mRNA was used to analyze the effect of leptin on the expression of selected genes and proteins involved in the cell cycle and apoptosis. OVCAR-3 cells were exposed to 2, 20, 40, and 100 ng/ml of leptin. Cell proliferation was determined using the alamarBlue cell viability test and flow cytometry. Apoptosis was measured using a cellular DNA fragmentation ELISA kit. The expression of selected cell cycle and apoptosis genes was evaluated by real-time PCR and confirmed by western blot. The stimulatory action of leptin on cell proliferation was observed as an increase in cells in the S and G2/M phases. Up-regulation of genes responsible for inducing cell proliferation and suppression of genes responsible for inhibition of proliferation were noted. Western blots revealed increased expression of cyclins D and A and inhibition of p21WAF1/CIP1 protein expression by leptin. Inhibition of DNA fragmentation was observed under all leptin doses. Suppression of genes involved in the extrinsic and intrinsic apoptotic pathway was observed. Western blots illustrated decreased Bad, TNFR1, and caspase 6 protein expression in response to leptin treatment. Leptin promotes ovarian cancer cell line growth by up-regulating genes and proteins responsible for inducing cell proliferation as well as down-regulating pro-apoptotic genes and proteins in apoptotic pathways. Results of this study warrant examining the relationship between the risk of ovarian cancer and elevated leptin levels in obese women.
Similar content being viewed by others

Introduction
Leptin is a multifunctional peptide hormone with wide-ranging biologic activities, including appetite regulation, bone formation, reproductive function, and angiogenesis. Circulating levels of leptin are strongly correlated to body fat content and are markedly elevated in obese compared with normal-weight individuals [1, 2]. Plasma leptin levels have been reported to be higher in overweight and obese women (37.7 ng/ml) than in normal-weight women (3.92–16.9 ng/ml) [2–4]. In some obese individuals, leptin levels can reach 100 ng/ml [5]. Interestingly, recent studies have demonstrated that leptin stimulates growth, migration, invasion, and angiogenesis in tumor cell models, suggesting that it can promote an aggressive cancer phenotype [6, 7]. Epidemiological studies have suggested a positive correlation between obesity and increased risk of different cancers, including ovarian cancer [8–10]. Several studies have addressed the possible role of leptin, the product of the obesity gene (Ob), in ovarian cancer development and progression [11–13]. A recent study by Uddin et al. [13] showed a significant association between leptin receptor (Ob-R) overexpression and poor progression-free survival in 59.2 % of epithelial ovarian cancers. In vitro studies have demonstrated that leptin induces proliferation in the ovarian cancer cell line BG-1 [11, 12], and it has been shown to inhibit apoptosis in ovarian epithelial cancer cell lines SKOV3 and MDAH2774 [13]. Accumulating evidence indicates that leptin has an effect on ovarian cancer growth, but information on its molecular mechanism with respect to regulation of the cell cycle and apoptosis is limited.
Based on the data of Choi et al. [11], who characterized both long (ObRb) and short (ObRt) isoforms of leptin mRNA, as well as our previous study [14] showing protein expression of both isoforms of the leptin receptor in the immortalized OVCAR-3 ovarian cancer cell line, we used this cell line to analyze the effect of leptin on the expression of selected genes involved in the cell cycle and apoptosis.
Materials and methods
Reagents
RPMI 1640 medium without phenol red, fetal bovine serum (FBS, heat-inactivated), penicillin, streptomycin, and trypsin EDTA were obtained from PAA Laboratories GmbH (Colbe, Germany). Human leptin, bovine serum albumin (BSA), and trypan blue were obtained from Sigma Chemical Co., (St. Louis, MO, USA).
Cell culture and treatments
The human ovarian epithelial carcinoma cell line OVCAR-3 was obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were routinely cultured in RPMI 1640 medium without phenol red supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin, and 10 % v/v heat-inactivated FBS in a humidified incubator with 5 % CO2 at 37 °C. The cells were switched to medium without serum 24 h before each experiment.
Cell proliferation
Cell proliferation was measured using the alamarBlue cell viability reagent (Invitrogen, Paisley, UK) according to the manufacturer’s instructions. The cells were seeded in 96-well culture plates at a density of 1.5 × 104 cell/well and then incubated in RPMI 1640 supplemented with 5 % FBS as a control medium or with four different doses (2, 20, 40, and 100 ng/ml) of leptin for 24, 48, and 72 h. The medium was changed daily, adding new medium and new test compounds. The alamarBlue stock solution was aseptically added to the wells after 24, 48, and 72 h of culture in amounts equal to 10 % of the incubation volume. Resazurin reduction was determined after 4 h of incubation by measuring the fluorescence at 560 nm (excitation)/590 nm (emission) using an FLx800 fluorescence microplate reader (BioTek, Winooski, VT, USA).
Flow cytometry analyses
The cells were seeded in 6-well culture plates at a density of 0.5 × 106 cell/well in RPMI 1640 supplemented with 5 % FBS as a control medium or with 40 ng/ml of leptin for 24 and 48 h. For analysis of cell proliferation, cell samples were stained with propidium iodide (PI). Briefly, after centrifugation (10 min at 1,500 rpm, 4 °C), the cell pellets were adjusted to 1 × 106 cell/ml and re-suspended in the PI staining buffer. The buffer contained 7.5 × 10−5 M PI, 10 mM Tris base, 10 mM NaCl, 700 U/l RNase, 0.1 ml/100 ml Nonidet P-40 (all from Sigma-Aldrich Chemical Co., St. Louis, MO, USA) and was adjusted to pH 8.0. The re-suspended cell pellets were kept in the buffer for 30 min in the dark at 4 °C. Samples were then analyzed in a FACScalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) to assess the cell cycle DNA profile. In total, 20,000 threshold events per sample were collected and analyzed based on their FL2 fluorescence (emission at 585 nm, excitation at 488 nm). The resulting data were analyzed using CellQuest Pro software (Becton-Dickinson). Percentages of cells present in the various phases of the cell cycle were calculated using Cell Quest by gating for the following cell populations: A (apoptotic cells; <2n), G1/G0 [cells in gap 1 (G1) and entering into the quiescent phase (G0); 2n], S (cells in the DNA synthesis phase; >2n), and G2 + M [cells in gap 2 (G2; 4n), and entering into the mitotic phase (M; <4n)].
Bromodeoxyuridine (BrdU) incorporation assay
DNA fragmentation was determined using the Cellular DNA Fragmentation ELISA Kit (Roche Applied Science, Mannheim, Germany). This assay is based on the quantitative detection of BrdU-labeled DNA fragments. After exposure to BrdU for 18 h, the cells were re-seeded in 96-well culture plates at 1.5 × 104 cell/well and then incubated in RPMI 1640 as a control medium or with leptin at concentrations of 2, 20, 40, and 100 ng/ml. After 24 h, DNA fragmentation was determined according to the manufacturer’s instructions. Absorbance values were measured spectrophotometrically at 450 nm using an ELx808 ELISA reader (BioTek).
Real-time PCR analysis
Cells were seeded in 6-well culture plates at 1 × 106 cell/well in RPMI 1640 supplemented with 5 % FBS (for cell cycle analysis) or without serum (for apoptosis analysis) as the control medium, or with 40 ng/ml of leptin for 24 h. The choice of this dose was based on previous cell proliferation experiments. Total RNA from OVCAR-3 cells was isolated using the High Pure RNA Isolation Kit (Roche Applied Science). RNA purity and quantity were determined spectrophotometrically according to optical densities at 260 and 280 nm. cDNA synthesis was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science) with a mixture of anchored-oligo(dT)18 primers and random hexamer primers according to the manufacturer’s protocol. Amplifications were performed in duplicate using the LightCycler 480 System (Roche Applied Science) and the Real-Time Ready Human Apoptosis Panel and Real-Time Ready Human Cell Cycle Regulation Panel (Roche Applied Science) in combination with the LightCycler 480 Probes Master (Roche Applied Science) according to the manufacturer’s instructions. PCR was performed in a final volume of 20 μl, including 50 ng cDNA per reaction. The PCR conditions were as follows: pre-incubation for 10 min at 95 °C, amplification for 45 cycles (10 s at 95 °C, 30 s at 60 °C, and 1 s at 72 °C). A positive control (to check for RNA degradation) and negative control (to detect residual genomic DNA) were run simultaneously with every assay according to the manufacturer’s protocol. The relative expression of the analyzed genes was normalized to seven reference genes in the panel using the E-Method from Roche Applied Science, which analyzes the amplification efficiency of target and reference genes using relative standards. These standards are serial dilutions of a single sample (for example, undiluted, 1:10, 1:100, and so on), where the concentration is expressed in relative units (for example, 1, 0.1, 0.01, and so on). Using dilutions to generate a standard curve, the E-Method avoids the time-consuming preparation of artificial or cloned standards and the determination of their absolute values [15].
Western blot analysis
The cells were seeded in 24-well culture plates at 1 × 105 cell/well in RPMI 1640 supplemented with 5 % FBS (for cell cycle analysis) or without serum (for apoptosis analysis) as the control medium, or with leptin at 40 ng/ml for 24 and 48 h. The cells were washed with ice-cold PBS and lysed in ice-cold buffer. The protein concentration of the cell lysate was determined by Bradford reagent (Bio-Rad Protein Assay, Bio-Rad Laboratories, Munchen, Germany). Protein (30 µg from each treatment group) was separated by 10 and 15 % SDS-PAGE and transferred to PVDF membranes using a Bio-Rad Mini-Protean 3 apparatus (Bio-Rad Laboratories). The blots were blocked for 2 h with 5 % w/v BSA and 0.1 % v/v Tween 20 in 0.02 M Tris Buffered Saline buffer (TBS). The blots were incubated overnight at 4 °C with antibodies specific for cyclin D1, cyclin A, p21 Waf1/Cip1, TNFR1, caspase 6, and Bad from Cell Signaling Technology (Danvers, MA, USA). Loading controls consisted of immunoblotting the same membranes with β-actin antibody (Sigma Chemical Co.,). After incubation with the primary antibody, the membranes were washed three times and incubated for 1 h with a horseradish peroxidase-conjugated secondary antibody from Cell Signaling Technology for cyclin D1, cyclin A, p21 Waf1/Cip1, TNFR1, caspase 6, and Bad (from DakoCytomation, Glostrup, Denmark) for β-actin. Immunopositive bands were visualized using Western Blotting Luminol Reagents (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and quantified using densitometry analysis (EasyDens, Cortex Nowa, Poland).
Statistical analysis
Data were expressed as mean ± SEM from four independent experiments performed in triplicate. Statistical analyses were performed using GraphPad Prism 5. Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference (HSD) multiple range test. A value of P < 0.05 was considered statistically significant.
Results
Cell proliferation (alamarBlue assay)
Growth of OVCAR-3 cells in response to various doses of leptin was tested for 24, 48, and 72 h. An increase in cell proliferation was noted under all leptin doses at 48 h of treatment (1.3-fold; P < 0.05; Fig. 1), with 2 and 20 ng/ml of leptin at 72 h of treatment (1.3-fold; P < 0.05; Fig. 1), and with 40 and 100 ng/ml of leptin at 72 h of treatment (1.6-fold; P < 0.001; Fig. 1). The sub-maximal leptin dose (40 ng/ml) was chosen for subsequent experiments.

Effects of leptin on OVCAR-3 cell proliferation. The cells were treated with leptin at 2, 20, 40, and 100 ng/ml for 24, 48, and 72 h and then analyzed using the alamarBlue assay. Values are mean ± SEM. *,***Significantly different from control at P < 0.05 and P < 0.001, respectively
Distribution in the cell cycle (flow cytometry)
Figure 2 presents the distribution of cells in the various phases of the cell cycle. In the controls, most OVCAR-3 cells were in the G0/G1 phase at any given point of time. At 48 h, significantly different percentages of cells were in the S phase (DNA content 2–4n; 13.16 ± 0.40 % for leptin-treated cells vs. 10.99 ± 0.28 % for the control) and G2 + M phase (4n; 22.76 ± 0.38 for leptin-treated cells vs. 20.08 ± 0.62 % for the control) (P < 0.05; Fig. 2a, b).

Effects of leptin (40 ng/ml) on cellular DNA content and cell proliferation. a Representative histograms as estimated by flow cytometry after propidium iodide staining. G0/G1 resting phase, in preparation for S; S DNA synthesis phase; G2 + M cells in gap 2 and entering into the mitotic phase; A apoptotic cells. Arrows indicate peaks of S and G2 + M cells. b Relative percentages of the cell population in S and G2/M phase are presented in the total gated cell numbers. Values are mean ± SEM. *Significantly different from the control at P < 0.05
Cell cycle gene expression analysis
A 1.2- to 1.9-fold induction of G1/S-specific cyclin D1 (CCND1), G2/mitosis-specific cyclin A2 (CCNA2), transcription factors E2F3 (E2F3), and proliferating cell nuclear antigen (PCNA) (all genes inducing cell proliferation) was measured in cells exposed to leptin. In parallel, 1.9- and 1.2-fold suppressions of cyclin-dependent kinase inhibitor p21 (P21CIP1) and p27 (P27KIP1), respectively, and a 1.2-fold suppression of growth arrest and DNA-damage-inducible protein GADD45 alpha (GADD45A) (genes inhibiting proliferation) were observed. No change was found in the levels of cell division protein kinases 1 (CDK1), 4 (CDK4), and 6 (CDK6), wee1-like protein kinase (WEE1), or transcription factors E2F1 (E2F1) or E2F2 (E2F2). We also noted no change in the levels of the S-phase regulatory protein cyclin E (CCNE2, CCNE2) or its partner protein CDK2 (CDK2), or in G2/mitotic-specific cyclin B1 (CCNB1) (P < 0.05, P < 0.001; Table 1).
Cell cycle protein expression analysis
The protein expression of CCND1, CCNA2, and P21CIP1, the most changed genes during the cell cycle, was analyzed by western blot. As with the genes’ expression, an increase in cyclin D1 (an S-phase-associated protein) and cyclin A2 (a G2/M-phase-associated protein) and a decrease in p21WAF1/CIP1 (inhibitor of S-phase-associated protein) expression following leptin treatment were noted (P < 0.01, P < 0.001; Fig. 3a–c).

Effects of leptin (40 ng/ml) on a cyclin D1 (36 kDa), b cyclin A (55 kDa), and c p21WAF1/CIP1 (21 kDa) protein expression in OVCAR-3 cells. Cells were treated with leptin for 24 and 48 h. β-actin (42 kDa) served as the loading control, and densitometry values of cyclin D1, cyclin A, and p21WAF1/CIP1 bands were normalized to β-actin. Signal intensities are expressed in arbitrary units. The values are mean ± SEM. **,***Significantly different from control at P < 0.01 and P < 0.001, respectively
Apoptosis analysis by DNA fragmentation
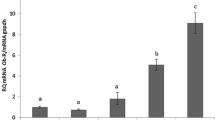
The statistically significant inhibition of DNA fragmentation to 84, 82, 75, and 74 % of control values was observed with leptin treatments of 2, 20, 40, and 100 ng/ml, respectively (P < 0.05, P < 0.01; Fig. 4).

Effects of leptin on DNA fragmentation in OVCAR-3 cells. Cells were treated with leptin at 2, 20, 40, and 100 ng/ml for 24 h and then analyzed with a Cellular DNA Fragmentation ELISA kit. RAU, relative absorbance units. Values are mean ± S.E.M. *,**Significantly different from control at P < 0.05 and P < 0.01, respectively
Apoptotic gene expression analysis
As shown in Table 2, leptin had no effect on FAS, CRADD, caspase 2 (CASP2), or caspase 8 (CASP8) mRNA levels, whereas we observed 1.4- to 1.8-fold suppression of TNFR1, FADD, and caspase 10 (CASP10). These results suggest a lack of activation of the Fas-dependent apoptotic pathway and inhibition of the TNFR apoptotic pathways. All of these genes are involved in the extrinsic apoptotic pathway. A 1.2-fold suppression of caspase 9 (CASP9) and 2.0-fold suppression of the gene expression of its partner protein Apaf-1 (APAF1) were noted. In addition, we measured a range of 1.3- to 2.2-fold suppression of caspases 3 (CASP3), 6 (CASP6), and 7 (CASP7), which are well-known effector caspases, and CAD and ICAD, which are responsible for DNA fragmentation (all of these genes are involved in the intrinsic apoptotic pathway). Leptin had no effect on BCL2, BCLW, BCLX, or MCL1 levels (pro-survival genes), whereas a range of 1.2- to 2.1-fold suppression of BAD, BAX, BAK1, BID, and BOK (pro-apoptotic genes) was observed. We also noted no change in the levels of the endonuclease G (ENDOG) genes involved in CAD-independent DNA fragmentation or in the tumor-suppressor gene p53 (TP53) (P < 0.05, P < 0.01, P < 0.001; Table 2).
Apoptotic protein expression analysis
The protein expression of BAD, TNFR1, and CASP6, the apoptotic genes that showed the largest changes in gene expression level, was analyzed by western blot. Leptin-induced inhibition of Bad (a component of the intrinsic pathway and a pro-apoptotic Bcl-2 family protein; P < 0.001; Fig. 5b), TNFR1 (a component of the extrinsic pathway, tumor necrosis factor receptor 1; P < 0.001; Fig. 5a), and caspase 6 protein (executor caspases; P < 0.001; Fig. 5c) expression was noted. The cleaved forms of caspase 6 were almost undetectable following leptin treatment.

Effects of leptin (40 ng/ml) on a Bad (23 kDa), b TNFR1 (55 kDa), and c caspase 6 (35, 15 kDa) protein expression in OVCAR-3 cells. Cells were treated with leptin for 24 and 48 h. β-actin (42 kDa) served as the loading control, and densitometry of TNFR1, Bad, and caspase 6 bands were normalized to β-actin. Signal intensities are expressed in arbitrary units. Values are mean ± SEM. ***Significantly different from control at P < 0.001
Discussion
We evaluated the effects of leptin on cell proliferation and apoptosis in the human ovarian cancer cell line OVCAR-3. Because we found a dose-dependent stimulatory effect on cell proliferation and an inhibitory effect on apoptosis, we expected that the gene products involved in these processes would also be affected.
Effects on cell proliferation
Using the alamarBlue test, which is based on the detection of cellular metabolic activity, we defined a dose-dependent (from 2 ng/ml to 100 ng/ml) stimulatory effect of leptin on OVCAR-3 cell proliferation. This observation is in agreement with Uddin et al. [13], who observed a dose-dependent (from 10 ng/ml) stimulatory effect of leptin on proliferation of the ovarian epithelial cancer cell line MDAH2774. On the other hand, Choi et al. [11] showed that leptin at 1 and 10 ng/ml doses has no effect on human ovarian cancer BG-1 cell proliferation, but stimulates cell proliferation at doses of 100 and 1,000 ng/ml.
Flow cytometry analysis showed that leptin promotes cell cycle progression, as demonstrated by an increased cell population in both the S and G2/M phases. Our results are consistent with Catalano et al. [16], who noted an increased number of Ishikawa human endometrial cancer cells in the S phase after 24 h of leptin treatment (100 and 1,000 ng/ml). In addition, Chen et al. [17] observed that 48-h treatment of ZR-75-1 breast cancer cells with leptin (100 ng/ml) increases the cell population in both S and G2/M phases.
Alterations of the mechanisms controlling cell cycle progression play a relevant role in the pathogenesis of different human neoplasias. Because the cell cycle is regulated by the coordinate action of cyclin-dependent kinases (cdks), specific cyclin proteins, and cdk inhibitors, we focused on the analysis of cell cycle-associated genes to gain insight into the mechanism of leptin’s proliferative action. Using real-time PCR, we found that leptin (40 ng/ml) impacts proliferation by up-regulating cell cycle progression genes and suppressing cell cycle inhibitor genes. We observed up-regulation of mRNAs for CCND1, CCNA2, PCNA, and E2F3 (genes responsible for inducing cell proliferation) and down-regulation of mRNAs for P21CIP1, P27KIP1, and GADD45A (genes responsible for inhibiting proliferation). Up-regulation of CCND1 and CCNA2 and down-regulation of P21CIP1, the most changed genes, were also observed at the protein level by western blot. Cyclin D1, an important cell cycle regulator, is required for completion of the G1/S transition in normal mammalian cells [18, 19] and is one of the most commonly affected proteins in abnormal states, such as cancer. Our results are consistent with previous studies showing similar leptin induction of cyclin D1 expression in human breast cancer [16, 17, 20], the colon cancer cell line HT-29 [21], and human hepatocarcinoma cells [22]. As cells progress into the S phase, cyclin A is expressed and becomes the primary cyclin associated with cdk2. Overexpression of cyclin A has been indicated in various human tumors, including breast [23], prostate [24], and ovarian cancer [25, 26]. To the best of our knowledge, this is the first report of leptin induction of cyclin A expression in human ovarian cancer cells. In the present study, we observed suppression of p21WAF1/CIP1, which acts as a universal inhibitor of cdks, and arrests the cell cycle at both G1/S and G2/M restriction points [27]. Our results are consistent with previous studies showing a similar decrease in p21WAF1/CIP1 expression under the influence of leptin in human breast cancer cells [17] and Ishikawa human endometrial cancer cells [16]. Furthermore, low levels of p21WAF1/CIP1 have been shown to be a marker of poor overall survival in ovarian cancer patients [28]. Down-regulation or inactivation of the cdk inhibitors, including p21CIP1, p27KIP1, and p16INK4a, which normally cause G1 arrest by binding to cyclin-cdk complexes, is often observed in a variety of human tumors, rendering the cell susceptible to uncontrolled extracellular proliferation signals [29].
To summarize, we suggest that leptin may promote progression from G1 to S phase by stimulating expression of cyclin D1 and inhibiting that of p21WAF1/CIP1, as well as progression to the S phase by stimulating the expression of cyclin A in an ovarian cancer cell line.
Action on cell apoptosis
A perturbation of the balance between cell proliferation and apoptosis has been suggested to play a significant role in mediating tumor progression. Surprisingly, data concerning the anti-apoptotic actions of leptin in ovarian cancer cells are limited. Herein, we observed that leptin inhibits OVCAR-3 cell apoptosis in a dose-dependent manner, as measured by DNA fragmentation. To our knowledge, there has been only one study addressing leptin in ovarian cancer cells [13], which showed that 100 ng/ml of leptin significantly prevents serum-starved apoptosis in the ovarian epithelial cancer cell lines SKOV-3 and MDAH2774. In previously published data, using physiologically relevant ovarian tissue, we illustrated the inhibitory action of leptin on cell apoptosis in pig ovarian follicles [30] and the developing corpus luteum [31]. There are also data showing an anti-apoptotic effect of leptin in breast cancer cell lines [7].
Cells undergo apoptosis through two major pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway. A hallmark of apoptosis is fragmentation of nuclear DNA. This process involves the caspase 3-dependent DNase CAD or can occur independently of caspase activity. In the present study, we found that 40 ng/ml of leptin acts by suppressing the expression of pro-apoptotic genes, such as TNFR1, FADD, CASP3, 6, 7, 9, and 10, CAD, ICAD, BAX, BAD, BAK1, BID, BOK, and APAF1. TNFR1, BAD, and CASP6 were the most down-regulated genes, and these changes were also observed at the protein level, as measured by western blot. Using a gene microarray, Perera et al. [32] observed that in MCF-7 cells, leptin (500 ng/ml) induces the expression of anti-apoptotic genes BCL2 and survivin and reduces the expression of other apoptotic genes (TRAF-interacting protein and TRADD) involved in the tumor necrosis factor (TNF)-induced apoptotic pathway. Their observation is partially consistent with our results, suggesting that leptin suppresses the TNF-induced extrinsic apoptosis pathway. Our data further showed suppression of TNFR1 at both the gene and protein expression levels. The leptin-induced reduction of Bax protein expression in T47-D breast cancer cells observed by Nkhata et al. [33], and in BAX gene expression observed here in OVCAR-3 cells, suggests that leptin also acts on the intrinsic apoptotic pathway. Down-regulation of TGFB1-induced Bax expression in hepatocarcinoma cells was described by Chen et al. [22]. Both apoptotic pathways lead to activation of the executioner caspases, caspase 3, 6, and 7, which are the main proteases degrading the cell. To the best of our knowledge, we are the first to show down-regulation of caspase 6 (executor caspase) at the gene and protein levels. Jiang et al. [34] showed leptin suppression of docetaxel-induced apoptosis in MCF-7 breast cancer cells via inhibition of caspase 9 activity, indirectly confirming our results. Similar to caspase 6, we observed inhibition of caspase 3 and caspase 7 mRNA expression by leptin, suggesting that leptin inhibits caspase-dependent apoptosis. Thus, we suggest that leptin prevents caspase-dependent apoptosis in OVCAR-3 cells by down-regulating pro-apoptotic proteins in both extrinsic (TNFR1) and intrinsic (BAD, caspase 6) apoptosis pathways.
In conclusion, our results are the first to demonstrate the molecular mechanism involved in the regulation of the cell cycle and apoptosis by leptin in an epithelial ovarian cancer cell line. The combination of proliferative and anti-apoptotic effects, by up-regulating genes and proteins responsible for inducing cell proliferation as well as down-regulating pro-apoptotic genes and proteins in the apoptotic pathways, clarifies the role of leptin signaling in the progression of ovarian cancer. Taking into consideration the limitations of in vitro studies, in vivo studies should be performed to confirm leptin’s contribution to ovarian cancer progression.
References
M.E. Grossmann, A. Ray, K.J. Nkhata, D.A. Malakhov, O.P. Rogozina, S. Dogan, M.P. Cleary, Obesity and breast cancer: status of leptin and adiponectin in pathological processes. Cancer Metastasis Rev. 29, 6416–6453 (2010)
M. Owecki, E. Nikisch, A. Miczke, D. Pupek-Musialik, J. Sowiński, Leptin, soluble leptin receptors, free leptin index, and their relationship with insulin resistance and BMI: high normal BMI is the threshold for serum leptin increase in humans. Horm. Metab. Res. 42, 585–589 (2010)
P.J. Havel, S. Kasim-Karakas, W. Mueller, P.R. Johnson, R.L. Gingerich, J.S. Stern, Relationship of plasma leptin to plasma insulin and adiposity in normal weight and overweight women: effects of dietary fat content and sustained weight loss. J. Clin. Endocrinol. Metab. 81, 4406–4413 (1996)
E. Kimura, K. Matsumoto, T. Samori, S. Kato, T. Kawahara, One-step enzyme-linked immunosorbent assay (ELISA) for measurement of serum free leptin. Clin. Chim. Acta 296, 45–57 (2000)
I. Knerr, D. Herzog, M. Rauh, W. Rascher, T. Horbach, Leptin and ghrelin expression in adipose tissues and serum levels in gastric banding patients. Eur. J. Clin. Invest. 36, 389–394 (2006)
C. Garofalo, E. Surmacz, Leptin and cancer. J. Cell. Physiol. 207, 12–22 (2006)
T. Jardé, S. Perrier, M.P. Vasson, F. Caldefie-Chézet, Molecular mechanisms of leptin and adiponectin in breast cancer. Eur. J. Cancer 47, 33–43 (2011)
C.M. Olsen, A.C. Green, D.C. Whiteman, S. Sadeghi, F. Kolahdooz, P.M. Webb, Obesity and the risk of epithelial ovarian cancer: a systematic review and meta-analysis. Eur. J. Cancer 43, 690–709 (2007)
L.J. Schouten, C. Rivera, D.J. Hunter, D. Spiegelman, H.O. Adami, A. Arslan, W.L. Beeson, P.A. van den Brandt, J.E. Buring, A.R. Folsom et al., Height, body mass index, and ovarian cancer: a pooled analysis of 12 cohort studies. Cancer Epidemiol. Biomarkers 17, 902–912 (2008)
M.F. Leitzmann, C. Koebnick, K.N. Danforth, L.A. Brinton, S.C. Moore, A.R. Hollenbeck, A. Schatzkin, J.V. Lacey Jr, Body mass index and risk of ovarian cancer. Cancer 115, 812–822 (2009)
J.H. Choi, S.H. Park, P.C. Leung, K.C. Choi, Expression of leptin receptors and potential effects of leptin on the cell growth and activation of mitogen-activated protein kinases in ovarian cancer cells. J. Clin. Endocrinol. Metab. 90, 207–210 (2005)
J.H. Choi, K.T. Lee, P.C. Leung, Estrogen receptor alpha pathway is involved in leptin-induced ovarian cancer cell growth. Carcinogenesis 32, 589–596 (2011)
S. Uddin, R. Bu, M. Ahmed, J. Abubaker, F. Al-Dayel, P. Bavi, K.S. Al-Kuraya, Overexpression of leptin receptor predicts an unfavorable outcome in Middle Eastern ovarian cancer. Mol. Cancer 8, 74 (2009)
A. Ptak, E.L. Gregoraszczuk, Bisphenol A induces leptin receptor expression, creating more binding sites for leptin, and activates the JAK/Stat, MAPK/ERK and PI3K/Akt signalling pathways in human ovarian cancer cell. Toxicol. Lett. 210, 332–337 (2012)
G. Tellmann, The E-method: a highly accurate technique for gene-expression analysis. Nat. Methods 3 (2006). doi:10.1038/nmeth894
S. Catalano, C. Giordano, P. Rizza, G. Gu, I. Barone, D. Bonofiglio, F. Giordano, R. Malivindi, D. Gaccione, M. Lanzino et al., Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J. Cell. Physiol. 218, 490–500 (2009)
C. Chen, Y.C. Chang, C.L. Liu, K.J. Chang, I.C. Guo, Leptin-induced growth of human ZR-75-1 breast cancer cells is associated with up-regulation of cyclin D1 and c-Myc and downregulation of tumor suppressor p53 and p21WAF1/CIP1. Breast Cancer Res. Treat. 98, 121–132 (2006)
M. Fu, C. Wang, Z. Li, T. Sakamaki, R.G. Pestell, Minireview: cyclin D1: normal and abnormal functions. Endocrinology 145, 5439–5447 (2004)
K.E. Knudsen, J.A. Diehl, C.A. Haiman, E.S. Knudsen, Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 25, 1620–1628 (2006)
N.K. Saxena, P.M. Vertino, F.A. Anania, D. Sharma, Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J. Biol. Chem. 282, 13316–13325 (2007)
P. Rouet-Benzineb, T. Aparicio, S. Guilmeau, C. Pouzet, V. Descatoire, M. Buyse, A. Bado, Leptin counteracts sodium butyrate-induced apoptosis in human colon cancer HT-29 cells via NF-kappaB signaling. J. Biol. Chem. 279, 16495–16502 (2004)
C. Chen, Y.C. Chang, C.L. Liu, T.P. Liu, K.J. Chang, I.C. Guo, Leptin induces proliferation and anti-apoptosis in human hepatocarcinoma cells by up-regulating cyclin D1 and down-regulating Bax via a Janus kinase 2-linked pathway. Endocr. Relat. Cancer 14, 513–529 (2007)
H. Kawamoto, H. Koizumi, T. Uchikoshi, Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am. J. Pathol. 150, 15–23 (1997)
R.D. Mashal, S. Lester, C. Corless, J.P. Richie, R. Chandra, K.J. Propert, A. Dutta, Expression of cell cycle-regulated proteins in prostate cancer. Cancer Res. 56, 4159–4163 (1996)
N. Barboule, V. Baldin, S. JOzan, S. Vidal, A. Valette, Increased level of p21 in human ovarian tumors is associated with increased expression of cdk2, cyclin A and PCNA. Int. J. Cancer 76, 891–896 (1998)
B.S. Yoon, Y.T. Kim, S. Kim, C.S. Lee, J.W. Kim, J.H. Kim, S.W. Kim, N.H. Cho, Prognostic value of nuclear DNA quantification and cyclin A expression in epithelial ovarian carcinoma. Eur. J. Obstet. Gynecol. Reprod. Biol. 136, 110–115 (2008)
Y. Xiong, G.J. Hannon, H. Zhang, D. Casso, R. Kobayashi, D. Beach, p21 is a universal inhibitor of cyclin kinases. Nature 366, 701–704 (1993)
A. Bali, P.M. O’Brien, L.S. Edwards, R.L. Sutherland, N.F. Hacker, S.M. Henshall, Cyclin D1, p53, and p21Waf1/Cip1 expression is predictive of poor clinical outcome in serous epithelial ovarian cancer. Clin. Cancer Res. 10, 5168–5177 (2004)
C.J. Sherr, J.M. Roberts, CDK inhibitors: positive and negative regulators of G1-phase progression. Gene. Dev. 13, 1501–1512 (1999)
E.L. Gregoraszczuk, A. Rak, A. Wójtowicz, A. Ptak, T. Wojciechowicz, K.W. Nowak, Gh and IGF-I increase leptin receptor expression in prepubertal pig ovaries: the role of leptin in steroid secretion and cell apoptosis. Acta Vet. Hung. 54, 413–426 (2006)
E.Ł. Gregoraszczuk, A. Ptak, In vitro effect of leptin on growth hormone (GH)- and insulin-like growth factor-I (IGF-I)-stimulated progesterone secretion and apoptosis in developing and mature corpora lutea of pig ovaries. J. Reprod. Dev. 51, 727–733 (2005)
C.N. Perera, H.G. Chin, N. Duru, I.G. Camarillo, Leptin-regulated gene expression in MCF-7 breast cancer cells: mechanistic insights into leptin-regulated mammary tumor growth and progression. J. Endocrinol. 199, 221–233 (2008)
K.J. Nkhata, A. Ray, T.F. Schuster, M.E. Grossmann, M.P. Cleary, Effects of adiponectin and leptin co-treatment on human breast cancer cell growth. Oncol. Rep. 21, 1611–1619 (2009)
H. Jiang, J. Yu, H. Guo, H. Song, S. Chen, Upregulation of survivin by leptin/STAT3 signaling in MCF-7 cells. Biochem. Biophys. Res. Commun. 368, 1–5 (2008)
Funding
This work was supported by the Polish Committee for Scientific Research from 2010 to 2013 as project 0050/B/P01/2010/38 (Poland).
Conflict of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the reported research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ptak, A., Kolaczkowska, E. & Gregoraszczuk, E.L. Leptin stimulation of cell cycle and inhibition of apoptosis gene and protein expression in OVCAR-3 ovarian cancer cells. Endocrine 43, 394–403 (2013). https://doi.org/10.1007/s12020-012-9788-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-012-9788-7