
Abstract
The vestibular system is a small bilateral structure located in the inner ear, known as the organ of balance and spatial orientation. It senses head orientation and motion, as well as body motion in the three dimensions of our environment. It is also involved in non-motor functions such as postural control of blood pressure. These regulations are mediated via anatomical projections from vestibular nuclei to brainstem autonomic centers and are involved in the maintenance of cardiovascular function via sympathetic nerves. Age-associated dysfunction of the vestibular organ contributes to an increased incidence of falls, whereas muscle atrophy, reduced physical activity, cellular aging, and gonadal deficiency contribute to bone loss. Recent studies in rodents suggest that vestibular dysfunction might also alter bone remodeling and mass more directly, by affecting the outflow of sympathetic nervous signals to the skeleton and other tissues. This review will summarize the findings supporting the influence of vestibular signals on bone homeostasis, and the potential clinical relevance of these findings.
Similar content being viewed by others

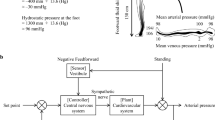
Introduction
The mammalian skeleton is kept mechanically sound in adults through the process of bone remodeling [1], which is driven by the coordinated and interdependent actions of osteoblasts, the bone-forming cells, and osteoclasts, the bone-resorbing cells [2]. Paracrine and cell-cell interactions couple the differentiation and activity of these two cell lineages. Thanks to the high vascularity and rich innervation of bones, hormones and neuroendocrine factors also link the process of bone remodeling to the function of other tissues, positioning the skeleton as an integral component of the complex homeostatic system of vertebrates [3, 4]. Bone cells receive cues from glands including the parathyroid gland (PTH); pancreas (insulin); and gonads (estradiol, testosterone, and inhibins) and from sympathetic nerves (norepinephrine). They also secrete hormones such as osteocalcin or FGF23 that regulate the function of other organs like the pancreas, kidneys, or the gonads [5–8, 9•, 10, 11], supporting the endocrine nature of the skeleton. In line with these inter-organ cross-talks, several centers within the central nervous system have been shown to control bone remodeling, and recent results in rats and mice suggest that the vestibular system, known for its role in regulating postural reflexes for balance, participates in the maintenance of bone mass, independent of its effects on locomotion. This review focuses on the regulation of adult bone homeostasis by the vestibular system and on the contribution of the sympathetic nervous system (SNS) to this process.
Bone Homeostasis Regulation by the Sympathetic Nervous System
The control of bone remodeling by the sympathetic nervous system (SNS) was discovered after the identification of leptin as an inhibitor of bone formation acting via a central relay [9•]. Leptin-deficient mice (the ob/ob mice) were indeed characterized by hypogonadism, hypercorticosteronemia, and increased vertebral cancellous bone mass, and leptin-responsive neurons in the ventromedial hypothalamus were shown to be required for normal bone remodeling. Four main observations suggested that sympathetic nerve activation, downstream of hypothalamic centers, was the mechanism by which such centers affect bone remodeling: (1) the high bone mass phenotype of leptin-deficient mice was accompanied by a low sympathetic tone [9•]; (2) electrical stimulation of ventromedial hypothalamic neurons [12] and leptin injection into the third ventricle area resulted in enhanced sympathetic tone [13]; (3) bones are innervated, as evidenced by immunological reactivity to various neuropeptides [14–19] and retrograde neuronal labeling [20]; and (4) the post-synaptic beta2-adrenergic receptors (β2AR) are expressed and functional in osteoblasts [6, 21, 22].
Based on these observations, different mouse genetic models and pharmacological approaches were then used to further address the contribution of sympathetic nerves to bone remodeling. Dopamine beta-hydroxylase (Dbh) is the gene encoding the enzyme that generates norepinephrine and epinephrine (the ligands for adrenergic receptors). Mice lacking Dbh, as well as mice lacking Adrb2 (the gene encoding the β2AR), displayed a high bone mass in adults [6, 23••]. In these models, there was an absence of the endocrine and metabolic changes observed in the ob/ob or Dbh−/− mice, thus excluding them as mediators of their bone phenotypes. Furthermore, leptin intracerebroventricular infusion failed to decrease bone mass in mice lacking the β2AR, demonstrating that the SNS, via the β2AR, mediates the central anti-osteogenic effect of leptin [6, 23••].
Drugs stimulating the β2ARs, such as the βAR agonists isoproterenol or clenbuterol, and drugs blocking β2AR signaling, such as the non-selective βAR blocker propranol, decrease and increase bone mass, respectively, in mice and rats [6, 24, 25•, 26]. In addition, lack of the norepinephrine transporter (Net), which functions in presynaptic neurons to reuptake norepinephrine and to control the duration and intensity of sympathetic signaling led to a low bone mass phenotype, suggesting that an endogenous homeostatic system exists to control or buffer the deleterious effect of sympathetic activation on the skeleton [27]. Collectively, these results provided multiple independent evidence that the SNS plays a critical role in the regulation of bone mass, at least in rodents. In humans, the observed association between the use of β-blockers and reduced fracture risk, and higher bone mineral density, further supported these results and their clinical relevance [27, 28] although this not a universal observation [29, 30]. The recording of higher SNS activity by microneurography at the peroneal nerve in postmenopausal women compared to premenopausal women was associated with reduced trabecular bone volume fraction and thickness by peripheral quantitative computed tomography (pQCT), suggesting here again that high sympathetic outflow leads to bone loss in humans [31].
Numerous conditions disturb the balance of bone remodeling, leading to a progressive decrease in bone mass, quality, architectural integrity, and mechanical properties. Aging is one such condition contributing to osteoporosis and bone fragility, independent of gonadal function. Oxidative stress is thought to be a main contributor to the deterioration of bone mass observed with aging [32, 33]. However, astronauts, who are relatively young and extremely fit individuals, also suffer bone loss. In such case, a reduction in dynamic load experienced by bone appears to be the main causal factor, as evidenced by human and animal studies in space and unloading studies on earth [34, 35, 36•]. While the contribution of mechanical signals is well-established, the involvement of the SNS in this process is now to be considered as an additional contributory factor. Indeed, anti-resorptives such as bisphosphonates, exercise regimens that mechanically load the skeleton, and nutritional supplementation countermeasures to prevent bone loss in space are still not fully effective, suggesting the existence of additional factors causing bone loss in weightlessness conditions [37–41]. What is especially interesting is that aged individuals and astronauts have at least three pathophysiological changes in common: a dysfunction of the vestibular system [42, 43•], an increase in SNS activity [44–46], and bone loss [42, 47]. The observations that vestibular dysfunction alter sympathetic outflow and that sympathetic nerve activity regulates bone remodeling begged the question as to whether the vestibular system had an influence on bone homeostasis.
The SNS is a Major Pathway for Vestibular Signal Outputs
The vestibular system is an integral part of the labyrinth that lies in the otic capsule in the petrous portion of the temporal bone. It is a sensory organ commonly known as the organ of balance and spatial orientation. Through three semicircular canals and two otolithic sensors (Fig. 1), it detects head orientation and motion, as well as body motion in the three dimensions. This system is studied in humans largely in the context of vestibular pathology like Menière’s disease, and more generally in all vestibular causes of vertigo, in order to elucidate the process of vestibular compensation and related pharmacological targets for balance recovery. Previous studies demonstrated its contribution to the regulation of posture, respiration, heart rate, and blood pressure in animals [48–51] and humans [52, 53], and via anatomic projections from vestibular nuclei to brainstem autonomic centers [54, 55]. All vestibular inputs transit via the vestibular nucleus to the brain through specific neuronal pathways, according to their function. For instance, oculomotor control and postural reflexes for balance involve the brainstem [56], whereas body and self-motion perception for balance and voluntary motor control involve cortical neurons [57, 58]. While high latency responses (>18 ms) are relayed to preganglionic sympathetic neurons through neurons in the caudal medullary raphe nuclei [59–61] and in the subretrofacial nuclei [61], short latency responses transit through the vestibulo-spinal axis in the lateral tegmental field [62] or through the subretrofacial neurons in the rostral ventrolateral medulla [63, 64]. Moreover, higher integrative centers, such as the hypothalamus [65–68], the nucleus of the solitary tract [55, 69], and the cerebellum [70] are also involved in the vestibulo-sympathetic responses [71, 72•, 73, 74], as demonstrated by the orthostatic hypotension observed in cats with bilateral vestibular destruction [75].

Model of the control of bone homeostasis by the vestibular system
Vestibular Outputs Participate to Bone Homeostasis Regulation Through the Sympathetic Nervous System
Retrospective studies highlighted a relationship between bone mineral density in aged patients and balance control [76, 77••]. From these studies, it was concluded that a reduction in balance control in aged individuals led to an increase in the risk of falls, and in turn, to an increase in bone fracture incidence. Further, it was suggested that balance disorders in aged individuals might be a result of loss of bone mineral density in the petrous bone surrounding the vestibular organ. However, the association between petrous bone density and vestibular function is not supported, and in the absence of additional data, the possibility that vestibular dysfunction may independently affect bone homeostasis and increase the rate of fracture upon aging was not considered.
This potential effect of vestibular dysfunction on bone density and homeostasis was prompted by the observation that sympathetic outflow is increased following alteration of vestibular signals in weightlessness, as noted above, and that sympathetic activation leads to bone loss. Levasseur et al. first reported a reduction in bone mineral density of the femoral metaphysis in rats after 1 month of chemically induced vestibular deficiency [78, 79]. This model used bilateral transtympanic sodium arsenilate injections to disturb vestibular function, causing vestibular deficiency that was accompanied with a series of abnormal spontaneous behaviors. These original results were confirmed in another study in rats, in which bone parameters were further assessed by uCT and histomorphometry [80•]. In addition, this study demonstrated that propranolol reduces the bone loss induced by transtympanic bilateral sodium arsenilate injections, supporting the contribution of sympathetic signaling to the effect of vestibular alterations on bone mass [80•].
Sodium arsenilate transtympanic injections are assumed to induce vestibular damage [81] and to promote degeneration of the eighth cranial nerve [82]. However, contrary to the high dose previously used, the low dose of sodium arsenilate used in studies examining bone loss did not induce changes in vestibular structure or nerve degeneration [83]. This observation suggested that low-dose sodium arsenilate modulates vestibular function, at least transiently and sufficiently to disturb bone homeostasis. In addition, these lesions did not reduce locomotor activity, and did not increase TNFα serum levels, thus excluding reduced mechanical loading and chronic systemic inflammation as mechanisms by which vestibular lesions cause bone loss [80•, 84, 85••]. This does not exclude the possibility that the alteration in vestibular function may change the “perception” of bone loading so that the threshold for initiating a modeling or remodeling response is increased.
This model of vestibular dysfunction was later adapted to mice with the purpose of using genetic loss of function models to further address the mechanism(s) involved in the interactions between vestibular neurons and bone cells. Here again, bilateral sodium arsenilate transtympanic injections induced bone loss in cancellous bone in femoral diaphyses but not in the vertebrae (L3-L4), as observed in rats with vestibular deficiency. This regionalized effect of vestibular deficiency on weight-bearing bones and on the trabecular bone compartment is reminiscent to the patterning of sympathetic nerve activity in response to various vestibular stimulation [73, 86•, 87–92] and suggests that central centers and peripheral sympathetic nerves innervate and control bone remodeling differentially among various skeletal elements. These results might also be relevant to data from Sample et al. [93], who showed evidence that load-induced response of a single bone is neuronally regulated and induce adaptive responses in other skeletal elements.
The cellular changes induced by vestibular lesions, i.e., an increase in osteoclast density and a reduction in osteoblast number were partly reminiscent to the phenotypes observed following isoproterenol treatment [6, 85••], supporting the hypothesis that vestibular deficiency induces bone loss, at least in part, via sympathetic nerve activation. This specific question was further addressed by the use of mice lacking the β2AR, globally or specifically on osteoblasts. Both mutant mice subjected to bilateral vestibular arsenilate injections were resistant to bone loss, supporting the notion that sympathetic nerves mediate the effect of vestibular deficiency on bone remodeling [85••]. The resistance of the osteoblast-specific β2AR-deficient mice also suggested that the osteoblast is the main target of this vestibulo-sympathetic axis and excluded the contribution of changes in blood flow or other mechanisms to bone loss induced by vestibular lesions.
Does Vestibular Aging Contribute to Osteoporosis?
Considering the retrospective studies mentioned above, linking balance control and fracture risk in aged individuals and the data from animal models, we propose the hypothesis that vestibular aging contributes to age-related bone loss. A vestibulo-sympathetic reflex attenuation with aging has indeed been well documented and correlates with degeneration of the vestibular system. First, in humans, the density of hair cells and sensorial cells of the vestibular system decreases with age [94, 95]. Hair cells themselves can also undergo morphological changes with age, such as cilia disarrangement, increased cilia fragility, and development of giant cilia [95, 96]. The neuronal structures relaying vestibular signals can be affected with time as well, as revealed by a decrease in Scarpa’s ganglion cell number [97] and a decline in the number of primary neurons of the human vestibular system [98] in both aged humans and rats [99–101]. A prospective trial conducted with 70 healthy individuals traced a time-course of vestibular information integration and led to the conclusion that vestibular information reached a peak at the age of 40–49 years old and then decreased. This observation was independent of gender influence and has been attributed to aging of the vestibular system and absence of correction [102]. Additionally, the age-related changes in vestibulo-ocular and vestibulo-spinal reflexes have also been attributed to morphological degeneration of the vestibular system (i.e., hair cells loss, changes in synapse morphology, electrophysiologic alterations) [103–106]. The absence or decrease of postural effect on breathing frequency in the elderly, as compared to young individuals, also demonstrated that aging impairs vestibular-mediated activation of ventilation in humans [107]. In addition, morphological changes associated with aging of the otolith organs are associated with altered vestibular-mediated activation of the cardiorespiratory system, as evidenced by the attenuation in muscle-sympathetic nerve responses in aged subjects [108]. Otolith stimulation elicits differential activation of sympathetic nerve activity and vascular responses to muscle and skin in humans [73, 86•, 87–92]. Head-down rotation in 40 healthy subjects did not change mean arterial pressure and heart rate in younger or older subjects but renal blood velocity and vascular conductance significantly decreased in young subjects whereas these parameters did not change in older subjects. These data led to the conclusion that vestibulo-sympathetic functions are attenuated in older humans [109].
Returning to the analogy of bone loss between aged subjects and astronauts, the absence of gravity in space triggers a reduction in motoneuron excitability by the otolithic system (gravity sensor of the vestibule) [110]. Similar to aged subjects, remodeling of synaptic connections in vestibular sensorial hair cells and an increase in the number and the size of synaptic knobs have been reported in adult rats during short-term space missions [111, 112]. The vestibulo-spinal and vestibulo-ocular reflexes are also altered in these conditions [113–115].
While unloading, radiation, and nutritional deficiencies are all thought to contribute to bone loss in space, it is not known if alterations in the vestibulo-sympathetic axis contribute to weightlessness-induced bone loss. Interestingly, some of the vestibular deficits or alterations observed in these conditions disappear with time (phenomenon of compensation), while others, including balance, eye movements in response to head rotations in the dark, navigational abilities, and spatial memory, remain permanently altered [116–120]. In mice, bilateral vestibular sodium arsenilate lesions induce a vestibular syndrome and bone loss after a month, but bone mass returns to normal 3 or 6 months after lesions [85••]. Although this result would suggest the existence of compensation and by extension, a transient and clinically irrelevant effect on bone in humans, it remains to be addressed whether or not the vestibular structural and functional changes induced by long-term weightlessness in space or by a single sodium arsenilate injection on earth in mice/rats share similarities.
Conclusions
The data reviewed herein suggest that deleterious alterations in vestibular function leading to increased sympathetic outflow upon aging may contribute to age-related bone loss. These data also suggest that a similar pathogenic mechanism is at play in younger individuals during weightlessness or other vestibular dysfunctions, in addition to the well-accepted bone unloading that occurs in space. Although two retrospective studies in aged patients with osteoporosis support this speculation [76, 77••], it remains to be further addressed in preclinical models and in humans. Although the vestibular system has defined neuronal projections to brainstem neurons that control sympathetic outflow, the precise central nervous system pathways and neurotransmitters specifically involved in the observed bone loss induced by vestibular alterations remain to be explored. These findings also have clinical implications for patients with a history of vestibular pathologies such as labyrinthectomy, antibiotic treatment (aminoglycosides and platinum-based chemotherapy), vestibular neuritis, or Ménière’s disease. Interestingly, autistic children also suffer from vestibular dysfunction [121, 122] and decreased bone mineral density, and present with higher fracture risk than age-matched controls [123, 124]. Alterations of the vestibulo-sympathetic axis may thus have many potential pathophysiological consequences on the skeleton, which need to be further investigated to better understand the complexity of bone homeostasis and to identify new therapies to improve bone mass.
Abbreviations
- SNS:
-
Sympathetic nervous system
- β2-AR:
-
Beta2-adrenergic receptors
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801.
Sims NA, Gooi JH. Bone remodeling: multiple cellular interactions required for coupling of bone formation and resorption. Semin Cell Dev Biol. 2008;19:444–51.
Elefteriou F, Campbell P, Ma Y. Control of bone remodeling by the peripheral sympathetic nervous system. Calcif Tissue Int. 2014;94:140–51.
Karsenty G, Ferron M. The contribution of bone to whole-organism physiology. Nature. 2012;481:314–20.
Ferron M, Wei J, Yoshizawa T, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296–308.
Yadav VK, Oury F, Suda N, et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138:976–89.
Oury F, Sumara G, Sumara O, et al. Endocrine regulation of male fertility by the skeleton. Cell. 2011;144:796–809.
Takeda S, Elefteriou F, Levasseur R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–17.
Ducy P, Amling M, Takeda S, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207. This publication brings the first evidence of a central control of bone remodeling and bone mass in mice.
Quarles LD. Evidence for a bone-kidney axis regulating phosphate homeostasis. J Clin Invest. 2003;112:642–6.
Lee NK, Sowa H, Hinoi E, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–69.
Perkins MN, Rothwell NJ, Stock MJ, et al. Activation of brown adipose tissue thermogenesis by the ventromedial hypothalamus. Nature. 1981;289:401–2.
Satoh N, Ogawa Y, Katsuura G, et al. Sympathetic activation of leptin via the ventromedial hypothalamus: leptin-induced increase in catecholamine secretion. Diabetes. 1999;48:1787–93.
Bjurholm A, Kreicbergs A, Brodin E, et al. Substance P- and CGRP-immunoreactive nerves in bone. Peptides. 1988;9:165–71.
Bjurholm A, Kreicbergs A, Terenius L, et al. Neuropeptide Y-, tyrosine hydroxylase- and vasoactive intestinal polypeptide-immunoreactive nerves in bone and surrounding tissues. J Auton Nerv Syst. 1988;25:119–25.
Goto T, Yamaza T, Kido MA, et al. Light- and electron-microscopic study of the distribution of axons containing substance P and the localization of neurokinin-1 receptor in bone. Cell Tissue Res. 1998;293:87–93.
Hill EL, Elde R. Distribution of CGRP-, VIP-, D beta H-, SP-, and NPY-immunoreactive nerves in the periosteum of the rat. Cell Tissue Res. 1991;264:469–80.
Hohmann EL, Elde RP, Rysavy JA. Innervation of periosteum and bone by sympathetic vasoactive intestinal peptide-containing nerve fibers. Science (New York, NY). 1986;232:868–71.
Sisask G, Bjurholm A, Ahmed M, et al. The development of autonomic innervation in bone and joints of the rat. J Auton Nerv Syst. 1996;59:27–33.
Dénes A, Boldogkoi Z, Uhereczky G, et al. Central autonomic control of the bone marrow: multisynaptic tract tracing by recombinant pseudorabies virus. Neuroscience. 2005;134:947–63.
Togari A, Arai M, Mizutani S, et al. Expression of mRNAs for neuropeptide receptors and beta-adrenergic receptors in human osteoblasts and human osteogenic sarcoma cells. Neurosci Lett. 1997;233:125–8.
Kellenberger S, Muller K, Richener H, et al. Formoterol and isoproterenol induce c-fos gene expression in osteoblast-like cells by activating beta2-adrenergic receptors. Bone. 1998;22:471–8.
Elefteriou F, Ahn JD, Takeda S, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–20. This study demonstrates the requirement of the peripheral sympathetic nervous system for normal bone remodeling in mice.
Bonnet N, Brunet-Imbault B, Arlettaz A, et al. Alteration of trabecular bone under chronic beta2 agonist’s treatment. Med Sci Sports Exerc. 2005;37:1493–501.
Bonnet N, Benhamou CL, Malaval L, et al. Low dose beta-blocker prevents ovariectomy-induced bone loss in rats without affecting heart functions. J Cell Physiol. 2008;217:819–27. Evaluation of the dose response of a non-selective beta-blocker on bone and heart functions in ovariectomized rats led to the conclusion that a low dose beta-blocker prevents bone loss without affecting heart functions.
Moore RE, Smith 2nd C, Bailey CS, et al. Characterization of beta-adrenergic receptors on rat and human osteoblast-like cells and demonstration that beta-receptor agonists can stimulate bone resorption in organ culture. Bone Miner. 1993;23:301–15.
Ma Y, Krueger JJ, Redmon SN, et al. Extracellular norepinephrine clearance by the norepinephrine transporter is required for skeletal homeostasis. J Biol Chem. 2013;288:30105–13.
Schlienger RG, Kraenzlin ME, Jick SS, et al. Use of beta-blockers and risk of fractures. JAMA: J Am Med Assoc. 2004;292:1326–32.
Rejnmark L, Vestergaard P, Kassem M, et al. Fracture risk in perimenopausal women treated with beta-blockers. Calcif Tissue Int. 2004;75:365–72.
Reid IR, Gamble GD, Grey AB, et al. beta-Blocker use, BMD, and fractures in the study of osteoporotic fractures. J Bone Miner Res. 2005;20:613–8.
Farr JN, Charkoudian N, Barnes JN, et al. Relationship of sympathetic activity to bone microstructure, turnover, and plasma osteopontin levels in women. J Clin Endocrinol Metab. 2012;97:4219–27.
Riggs BL, Khosla S, Melton 3rd LJ. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002;23:279–302.
Manolagas SC, Kousteni S, Jilka RL. Sex steroids and bone. Recent Prog Horm Res. 2002;57:385–409.
Sakai A, Nakamura T. Changes in trabecular bone turnover and bone marrow cell development in tail-suspended mice. J Musculoskelet Neuronal Interact. 2001;1:387–92.
Ehara Y, Yamaguchi M. Histomorphological confirmation of bone loss in the femoral-metaphyseal tissues of rats with skeletal unloading. Res Exp Med (Berl). 1996;196:163–70.
LeBlanc AD, Spector ER, Evans HJ, et al. Skeletal responses to space flight and the bed rest analog: a review. J Musculoskelet Neuronal Interact. 2007;7:33–47. This review summarizes four decades of human skeletal data from space programs and ground-based analog (bed rest) studies and discusses possible countermeasures to bone loss.
Cavanagh PR, Licata AA, Rice AJ. Exercise and pharmacological countermeasures for bone loss during long-duration space flight. Gravit Space Biol Bull. 2005;18:39–58.
Iwamoto J, Takeda T, Sato Y. Interventions to prevent bone loss in astronauts during space flight. Keio J Med. 2005;54:55–9.
Sibonga JD, Evans HJ, Sung HG, et al. Recovery of spaceflight-induced bone loss: bone mineral density after long-duration missions as fitted with an exponential function. Bone. 2007;41:973–8.
Smith SM, Heer MA, Shackelford LC, et al. Benefits for bone from resistance exercise and nutrition in long-duration spaceflight: Evidence from biochemistry and densitometry. J Bone Miner Res. 2012;27:1896–906.
Leblanc A, Matsumoto T, Jones J, et al. Bisphosphonates as a supplement to exercise to protect bone during long-duration spaceflight. Osteoporos Int. 2013;24:2105–14.
Tavassoli M. Medical problems of space flight. Am J Med. 1986;81:850–4.
Hsieh L-C, Lin H-C, Lee G-S. Aging of vestibular function evaluated using correlational vestibular autorotation test. Clin Interv Aging. 2014;9:1463–9. Here the authors show that the function of the visual-vestibulo-ocular reflex and of the vestibulo-ocular reflex declines with aging in humans.
Narkiewicz K, Phillips BG, Kato M, et al. Gender-selective interaction between aging, blood pressure, and sympathetic nerve activity. Hypertension. 2005;45:522–5.
Mano T. Autonomic neural functions in space. Curr Pharm Biotechnol. 2005;6:319–24.
Norsk P, Christensen NJ. The paradox of systemic vasodilatation and sympathetic nervous stimulation in space. Respir Physiol Neurobiol. 2009;169(1):S26–9.
Emkey GR, Epstein S. Secondary osteoporosis: pathophysiology & diagnosis. Best Pract Res Clin Endocrinol Metab. 2014;28:911–35.
Barmack NH. Central vestibular system: vestibular nuclei and posterior cerebellum. Brain Res Bull. 2003;60:511–41.
Yates BJ. The vestibular system and cardiovascular responses to altered gravity. Am J Physiol. 2004;286:R22.
Etard O, Reber A, Quarck G, et al. Vestibular control on blood pressure during parabolic flights in awake rats. Neuroreport. 2004;15:2357–60.
Abe C, Kawada T, Sugimachi M, et al. Interaction between vestibulo-cardiovascular reflex and arterial baroreflex during postural change in rats. J Appl Physiol. 2011;111:1614–21.
Normand H, Etard O, Denise P. Otolithic and tonic neck receptors control of limb blood flow in humans. J Appl Physiol (1985). 1997;82:1734–8.
Herault S, Tobal N, Normand H, et al. Effect of human head flexion on the control of peripheral blood flow in microgravity and in 1 g. Eur J Appl Physiol. 2002;87:296–303.
Yates BJ, Siniaia MS, Miller AD. Descending pathways necessary for vestibular influences on sympathetic and inspiratory outflow. Am J Physiol. 1995;268:R1381–5.
Cai Y-L, Ma W-L, Wang J-Q, et al. Excitatory pathways from the vestibular nuclei to the NTS and the PBN and indirect vestibulo-cardiovascular pathway from the vestibular nuclei to the RVLM relayed by the NTS. Brain Res. 2008;1240:96–104.
Tanguy S, Quarck G, Etard O, et al. Vestibulo-ocular reflex and motion sickness in figure skaters. Eur J Appl Physiol. 2008;104:1031–7.
Cullen KE. The neural encoding of self-generated and externally applied movement: implications for the perception of self-motion and spatial memory. Front Integr Neurosci. 2014;7:108.
DeAngelis GC, Angelaki DE. The neural bases of multisensory processes. 2012.
Morrison SF, Gebber GL. Classification of raphe neurons with cardiac-related activity. Am J Physiol. 1982;243:R49–59.
Yates BJ, Yamagata Y. Convergence of cardiovascular and vestibular inputs on neurons in the medullary paramedian reticular formation. Brain Res. 1990;513:166–70.
Yates BJ, Goto T, Bolton PS. Responses of neurons in the caudal medullary raphe nuclei of the cat to stimulation of the vestibular nerve. Exp Brain Res. 1992;89:323–32.
Barman SM, Gebber GL. Lateral tegmental field neurons of cat medulla: a source of basal activity of ventrolateral medullospinal sympathoexcitatory neurons. J Neurophysiol. 1987;57:1410–24.
Dampney RA, Goodchild AK, McAllen RM. Vasomotor control by subretrofacial neurones in the rostral ventrolateral medulla. Can J Physiol Pharmacol. 1987;65:1572–9.
Van Bockstaele EJ, Pieribone VA, Aston-Jones G. Diverse afferents converge on the nucleus paragigantocellularis in the rat ventrolateral medulla: retrograde and anterograde tracing studies. J Comp Neurol. 1989;290:561–84.
Herbert H, Moga MM, Saper CB. Connections of the parabrachial nucleus with the nucleus of the solitary tract and the medullary reticular formation in the rat. J Comp Neurol. 1990;293:540–80.
Balaban CD, Porter JD. Neuroanatomic substrates for vestibulo-autonomic interactions. J Vestib Res. 1998;8:7–16.
Cavdar S, San T, Aker R, et al. Cerebellar connections to the dorsomedial and posterior nuclei of the hypothalamus in the rat. J Anat. 2001;198:37–45.
Horowitz SS, Blanchard J, Morin LP. Medial vestibular connections with the hypocretin (orexin) system. J Comp Neurol. 2005;487:127–46.
Spyer KM. Neural organisation and control of the baroreceptor reflex. Rev Physiol Biochem Pharmacol. 1981;88:24–124.
Ishikawa T, Miyazawa T. Sympathetic responses evoked by vestibular stimulation and their interactions with somato-sympathetic reflexes. J Auton Nerv Syst. 1980;1:243–54.
Kerman IA, Yates BJ. Regional and functional differences in the distribution of vestibulosympathetic reflexes. Am J Physiol. 1998;275:R824–835.
Yates BJ, Miller AD. Physiological evidence that the vestibular system participates in autonomic and respiratory control. J Vestib Res. 1998;8:17–25. In this manuscript the authors show that body movements induce a vestibular response that aims to offset orthostatic hypotension by acting on the autonomic and respiratory systems in cats.
Kerman IA, Emanuel BA, Yates BJ. Vestibular stimulation leads to distinct hemodynamic patterning. Am J Physiol Regul Integr Comp Physiol. 2000;279:R118–25.
Kasumacic N, Glover JC, Perreault M-C. Vestibular-mediated synaptic inputs and pathways to sympathetic preganglionic neurons in the neonatal mouse. J Physiol. 2012;590:5809–26.
Mori RL, Cotter LA, Arendt HE, et al. Effects of bilateral vestibular nucleus lesions on cardiovascular regulation in conscious cats. J Appl Physiol. 2005;98:526–33.
Radaei F, Gharibzadeh S. Relationship between bone mineral density and balance disorders in osteoporotic patients. Front Bioeng Biotechnol. 2013;1:5.
Mendy A, Vieira ER, Albatineh AN, et al. Low bone mineral density is associated with balance and hearing impairments. Ann Epidemiol. 2014;24:58–62. In this retrospective study, the authors identified the association between low bone mineral density and balance in older adults and proposed that demineralization of the temporal bone, which contains the vestibular organ, leads to balance and hearing impairments.
Levasseur R, Sabatier JP, Etard O, et al. Labyrinthectomy decreases bone mineral density in the femoral metaphysis in rats. J Vestib Res. 2004;14:361–5.
Hunt MA, Miller SW, Nielson HC, et al. Intratympanic injection of sodium arsanilate (atoxyl) solution results in postural changes consistent with changes described for labyrinthectomized rats. Behav Neurosci. 1987;101:427–8.
Vignaux G, Besnard S, Ndong J, et al. Bone remodeling is regulated by inner ear vestibular signals. J Bone Miner Res. 2013;28:2136–44. This work identifies the vestibular system as a regulator of bone remodeling and bone mass in rats.
Anniko M, Wersäll J. Experimentally (atoxyl) induced ampullar degeneration and damage to the maculae utriculi. Acta Otolaryngol. 1977;83:429–40.
Andersson L, Ulfendahl M, Tham R. A method for studying the effects of neurochemicals on long-term compensation in unilaterally labyrinthectomized rats. J Neural Transplant Plast. 1997;6:105–13.
Vignaux G, Chabbert C, Gaboyard-Niay S, et al. Evaluation of the chemical model of vestibular lesions induced by arsanilate in rats. Toxicol Appl Pharmacol. 2012;258:61–71.
Ossenkopp KP, Prkacin A, Hargreaves EL. Sodium arsanilate-induced vestibular dysfunction in rats: effects on open-field behavior and spontaneous activity in the automated digiscan monitoring system. Pharmacol Biochem Behav. 1990;36:875–81.
Vignaux G, Ndong J, Perrien D, et al. Inner ear vestibular signals regulate bone remodeling via the sympathetic nervous system. J Bone Miner Res 2014. This study shows that the process of bone remodeling has a vestibulosympathetic regulatory component in mice, suggesting that vestibular system pathologies might cause bone fragility.
Kerman IA, McAllen RM, Yates BJ. Patterning of sympathetic nerve activity in response to vestibular stimulation. Brain Res Bull. 2000;53:11–6. This review of animal studies highlights the patterning of sympathetic response after vestibular stimulation, which is interestingly similar to the pattern of bone loss observed with aging or weightlessness.
Cui J, Mukai C, Iwase S, et al. Response to vestibular stimulation of sympathetic outflow to muscle in humans. J Auton Nerv Syst. 1997;66:154–62.
Ray CA, Hume KM, Steele SL. Sympathetic nerve activity during natural stimulation of horizontal semicircular canals in humans. Am J Physiol. 1998;275:R1274–8.
Shortt TL, Ray CA. Sympathetic and vascular responses to head-down neck flexion in humans. Am J Physiol. 1997;272:H1780–4.
Kaufmann H, Biaggioni I, Voustianiouk A, et al. Vestibular control of sympathetic activity. An otolith-sympathetic reflex in humans. Exp Brain Res. 2002;143:463–9.
Voustianiouk A, Kaufmann H, Diedrich A, et al. Electrical activation of the human vestibulosympathetic reflex. Exp Brain Res. 2006;171:251–61.
Bolton PS, Wardman DL, Macefield VG. Absence of short-term vestibular modulation of muscle sympathetic outflow, assessed by brief galvanic vestibular stimulation in awake human subjects. Exp Brain Res. 2004;154:39–43.
Sample SJ, Behan M, Smith L, et al. Functional adaptation to loading of a single bone is neuronally regulated and involves multiple bones. J Bone Miner Res. 2008;23:1372–81.
Rauch SD, Velázquez-Villaseñor L, Dimitri PS, et al. Decreasing hair cell counts in aging humans. Ann N Y Acad Sci. 2001;942:220–7.
Merchant SN, Velázquez-Villaseñor L, Tsuji K, et al. Temporal bone studies of the human peripheral vestibular system. Normative vestibular hair cell data. Ann Otol Rhinol Laryngol Suppl. 2000;181:3–13.
Rosenhall U, Rubin W. Degenerative changes in the human vestibular sensory epithelia. Acta Otolaryngol. 1975;79:67–80.
Velázquez-Villaseñor L, Merchant SN, Tsuji K, et al. Temporal bone studies of the human pe7ripheral vestibular system. Normative Scarpa’s ganglion cell data. Ann Otol Rhinol Laryngol Suppl. 2000;181:14–9.
Park JJ, Tang Y, Lopez I, et al. Age-related change in the number of neurons in the human vestibular ganglion. J Comp Neurol. 2001;431:437–43.
Nakayama M, Helfert RH, Konrad HR, et al. Scanning electron microscopic evaluation of age-related changes in the rat vestibular epithelium. Otolaryngol Head Neck Surg. 1994;111:799–806.
Sturrock RR. Age related changes in neuron number in the mouse lateral vestibular nucleus. J Anat. 1989;166:227–32.
Lopez I, Honrubia V, Baloh RW. Aging and the human vestibular nucleus. J Vestib Res. 1997;7:77–85.
Faraldo-García A, Santos-Pérez S, Crujeiras-Casais R, et al. Influence of age and gender in the sensory analysis of balance control. Eur Arch Otorhinolaryngol. 2012;269:673–7.
Chang C-M, Young Y-H, Cheng P-W. Age-related changes in ocular vestibular-evoked myogenic potentials via galvanic vestibular stimulation and bone-conducted vibration modes. Acta Otolaryngol. 2012;132:1295–300.
Sloane PD, Baloh RW, Honrubia V. The vestibular system in the elderly: clinical implications. Am J Otolaryngol. 1989;10:422–9.
Hirvonen TP, Aalto H, Pyykkö I, et al. Changes in vestibulo-ocular reflex of elderly people. Acta Otolaryngol Suppl. 1997;529:108–10.
Furman JM, Redfern MS. Effect of aging on the otolith-ocular reflex. J Vestib Res. 2001;11:91–103.
Kuipers NT, Sauder CL, Ray CA. Aging attenuates the vestibulorespiratory reflex in humans. J Physiol. 2003;548:955–61.
Ray CA, Monahan KD. Aging attenuates the vestibulosympathetic reflex in humans. Circulation. 2002;105:956–61.
Sauder CL, Conboy EE, Chin-Sang SA, et al. Otolithic activation on visceral circulation in humans: effect of aging. Am J Physiol Renal Physiol. 2008;295:F1166–9.
Reschke MF, Anderson DJ, Homick JL. Vestibulospinal reflexes as a function of microgravity. Science. 1984;225:212–4.
Ross MD. Morphological changes in rat vestibular system following weightlessness. J Vestib Res. 1993;3:241–51.
Ross MD, Tomko DL. Effect of gravity on vestibular neural development. Brain Res Brain Res Rev. 1998;28:44–51.
Dai M, McGarvie L, Kozlovskaya I, et al. Effects of spaceflight on ocular counter rolling and the spatial orientation of the vestibular system. Exp Brain Res. 1994;102:45–56.
Thornton WE, Uri JJ, Moore T, et al. Studies of the horizontal vestibulo-ocular reflex in spaceflight. Arch Otolaryngol Head Neck Surg. 1989;115:943–9.
Vogel H, Kass JR. European vestibular experiments on the Spacelab-1 mission: 7. ocular counter rolling measurements pre- and post-flight. Exp Brain Res. 1986;64:284–90.
Thomson DB, Inglis JT, Schor RH, et al. Bilateral labyrinthectomy in the cat: motor behaviour and quiet stance parameters. Exp Brain Res. 1991;85:364–72.
Stapley PJ, Ting LH, Kuifu C, et al. Bilateral vestibular loss leads to active destabilization of balance during voluntary head turns in the standing cat. J Neurophysiol. 2006;95:3783–97.
Barmack NH, Pettorossi VE, Erickson RG. The influence of bilateral labyrinthectomy on horizontal and vertical optokinetic reflexes in the rabbit. Brain Res. 1980;196:520–4.
Waespe W, Wolfensberger M. Optokinetic nystagmus (OKN) and optokinetic after-responses after bilateral vestibular neurectomy in the monkey. Exp Brain Res. 1985;60:263–9.
Baek JH, Zheng Y, Darlington CL, et al. Evidence that spatial memory deficits following bilateral vestibular deafferentation in rats are probably permanent. Neurobiol Learn Mem. 2010;94:402–13.
Siaperas P, Ring HA, McAllister CJ, et al. Atypical movement performance and sensory integration in Asperger’s syndrome. J Autism Dev Disord. 2012;42:718–25.
Molloy CA, Dietrich KN, Bhattacharya A. Postural stability in children with autism spectrum disorder. J Autism Dev Disord. 2003;33:643–52.
Neumeyer AM, Gates A, Ferrone C, et al. Bone density in peripubertal boys with autism spectrum disorders. J Autism Dev Disord. 2013;43:1623–9.
Neumeyer AM, O’Rourke JA, Massa A, et al. Brief report: bone fractures in children and adults with autism spectrum disorders. J. Autism Dev. Disord. 2014.
Acknowledgments
The authors would like to thank the National Aeronautics and Space Administration through grant NNX12AL35G (FE), the National Space Biomedical Research Institute (GV, NASA NCC 9–58), the European Union’s Seventh Framework Programme FP7/2007–2013/ through REA grant #318980, and the Centre National d’Etudes Spatiales (CNES, Grant #715) for their support of this work. We thank Dr. D. Perrien (VCBB) for critical reading of the manuscript.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
G Vignaux, S Besnard, P Denise, and F Elefteriou all declare no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by the authors involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Skeletal Biology and Regulation
Rights and permissions
About this article

Cite this article
Vignaux, G., Besnard, S., Denise, P. et al. The Vestibular System: A Newly Identified Regulator of Bone Homeostasis Acting Through the Sympathetic Nervous System. Curr Osteoporos Rep 13, 198–205 (2015). https://doi.org/10.1007/s11914-015-0271-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-015-0271-2