
Abstract
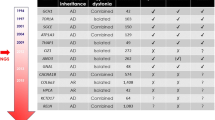
Dystonia, a common and genetically heterogeneous neurological disorder, was recently defined as “a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both.” Via the application of whole-exome sequencing, the genetic landscape of dystonia and closely related movement disorders is becoming exposed. In particular, several “novel” genetic causes have been causally associated with dystonia or dystonia-related disorders over the past 2 years. These genes include PRRT2 (DYT10), CIZ1 (DYT23), ANO3 (DYT24), GNAL (DYT25), and TUBB4A (DYT4). Despite these advances, major gaps remain in identifying the genetic origins for most cases of adult-onset isolated dystonia. Furthermore, model systems are needed to study the biology of PRRT2, CIZ1, ANO3, Gαolf, and TUBB4A in the context of dystonia. This review focuses on these recent additions to the family of dystonia genes, genotype–phenotype correlations, and possible cellular contributions of the encoded proteins to the development of dystonia.

Similar content being viewed by others

References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
LeDoux MS. The genetics of dystonias. Adv Genet. 2012;79:35–85. This is an encyclopedic review of dystonia genetics.
Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863–73.
LeDoux MS. Dystonia: phenomenology. Parkinsonism Relat Disord. 2012;18 Suppl 1:S162–4.
Fahn S. Classification of movement disorders. Mov Disord. 2011;26:94–57.
Ozelius LJ, Hewett JW, Page CE, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–8.
Hersheson J, Mencacci NE, Davis M, et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann Neurol. 2013;73:546–53. The authors used linkage analysis and whole-exome sequencing to identify a TUBB4A mutation in the “whispering dysphonia” pedigree.
Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236–42.
Fuchs T, Gavarini S, Saunders-Pullman R, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–8.
Rainier S, Thomas D, Tokarz D, et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch Neurol. 2004;61:1025–9.
Weber YG, Storch A, Wuttke TV, et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. 2008;118:2157–68.
Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43:1252–5.
Zimprich A, Grabowski M, Asmus F, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29:66–9.
Xiao J, Uitti RJ, Zhao Y, et al. Mutations in CIZ1 cause adult onset primary cervical dystonia. Ann Neurol. 2012;71:458–69. With use of linkage analysis and whole-exome sequencing, a pathogenic variant in CIZ1 was identified as the first cause of familial cervical dystonia. This work pointed out the potential role of the G 1 /S checkpoint pathway in the pathogenesis of primary dystonia.
Charlesworth G, Plagnol V, Holmstrom KM, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. 2012;91:1041–50. Mutations in ANO3 were identified as a potential cause of adult-onset dystonia, mainly cervical and segmental dystonia with dystonic tremors. Next-generation targeted sequencing was used to screen patients with familial and sporadic dystonia for SVs in ANO3.
Fuchs T, Saunders-Pullman R, Masuho I, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet. 2013;45:88–92. Fuchs et al. used whole-exome sequencing to identify GNAL mutations in relatively small pedigrees with mainly adult-onset cervical and segmental dystonia. Screening of primary dystonia pedigrees exposed additional putatively pathogenic variants. Impaired function of several of the mutations was shown by bioluminescence resonance energy transfer assays.
Ludecke B, Dworniczak B, Bartholome K. A point mutation in the tyrosine hydroxylase gene associated with Segawa's syndrome. Hum Genet. 1995;95:123–5.
Bonafe L, Thony B, Penzien JM, et al. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet. 2001;69:269–77.
Camargos S, Scholz S, Simon-Sanchez J, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7:207–15.
Nolte D, Niemann S, Muller U. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci U S A. 2003;100:10347–52.
Vemula SR, Puschmann A, Xiao J, et al. Role of Gα(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet. 2013;22:2510–9. Whole-exome sequencing identified a GNAL missense mutation (V228F) in an African-American pedigree with clinical phenotypes that include cervical, laryngeal ,and hand–forearm dystonia. Screening of 760 subjects with familial or sporadic primary dystonia identified three Caucasian pedigrees with GNAL mutations. Immunohistochemical studies showed that the encoded protein, Gα olf , is highly expressed in striatum and cerebellar Purkinje cells, and colocalized with corticotropin-releasing hormone receptors in Purkinje cells.
Lohmann K, Wilcox RA, Winkler S, et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol. 2013;73:537–45. The large pedigree was adequately powered for generation of a multipoint logarithm of odds score of 5.3. Then, whole-exome sequencing identified a missense mutation in TUBB4A as the cause of DYT4. Sequencing of TUBB4A in 394 unrelated dystonia patients identified one missense variant possibly associated with a case of segmental dystonia with spasmodic dysphonia.
LeDoux MS, Xiao J, Rudzinska M, et al. Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat Disord. 2012;18:414–25. This is a detailed and comprehensive analysis of coding and noncoding variants in THAP1 and exacting statistical analysis of THAP1 genotype–phenotype correlations.
Xiao J, Bastian RW, Perlmutter JS, et al. High-throughput mutational analysis of TOR1A in primary dystonia. BMC Med Genet. 2009;10:24.
Parker N. Hereditary whispering dysphonia. J Neurol Neurosurg Psychiatry. 1985;48:218–24.
Wilcox RA, Winkler S, Lohmann K, et al. Whispering dysphonia in an Australian family (DYT4): a clinical and genetic reappraisal. Mov Disord. 2011;26:2404–8.
Leandro-Garcia LJ, Leskela S, Landa I, et al. Tumoral and tissue-specific expression of the major human β-tubulin isotypes. Cytoskeleton (Hoboken). 2010;67:214–23.
Breuss M, Heng JI, Poirier K, et al. Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012;2:1554–62.
Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. 2013;92:767–73.
Blumkin L, Halevy A, Ben-Ami-Raichman D, et al. Expansion of the spectrum of TUBB4A-related disorders: a new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics. 2014;15:107–13.
Vemula SR, Xiao J, Bastian RW, et al. Pathogenic variants in TUBB4A are not found in primary dystonia. Neurology. 2014;82:1227–30. High-resolution melting and Sanger sequencing excluded TUBB4A coding region mutations in 575 subjects with primary laryngeal, segmental, or generalized dystonia.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–75.
Muller U, Steinberger D, Nemeth AH. Clinical and molecular genetics of primary dystonias. Neurogenetics. 1998;1:165–77.
Hedera P, Xiao J, Puschmann A, et al. Novel PRRT2 mutation in an African-American family with paroxysmal kinesigenic dyskinesia. BMC Neurol. 2012;12:93.
Caraballo R, Pavek S, Lemainque A, et al. Linkage of benign familial infantile convulsions to chromosome 16p12-q12 suggests allelism to the infantile convulsions and choreoathetosis syndrome. Am J Hum Genet. 2001;68:788–94.
Heron SE, Dibbens LM. Role of PRRT2 in common paroxysmal neurological disorders: a gene with remarkable pleiotropy. J Med Genet. 2013;50:133–9.
Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–68.
Uitti RJ, Maraganore DM. Adult onset familial cervical dystonia: report of a family including monozygotic twins. Mov Disord. 1993;8:489–94.
Ma L, Chen R, Wang L, et al. No mutations in CIZ1 in twelve adult-onset primary cervical dystonia families. Mov Disord. 2013;28:1899–901.
LeDoux MS. Exome sequencing for gene discovery: time does not stand still. Ann Neurol. 2012;72:628–9.
Zhao Y, Xiao J, Gong S, et al. Neural expression of the transcription factor THAP1 during development in rat. Neuroscience. 2013;231:282–95.
Xiao J, Gong S, Zhao Y, et al. Developmental expression of rat torsinA transcript and protein. Brain Res Dev Brain Res. 2004;152:47–60.
Ledoux MS. Non-Parkinson movement disorders: five new things. Neurol Clin Pract. 2013;3:22–9.
Munchau A, Valente EM, Davis MB, et al. A Yorkshire family with adult-onset cranio-cervical primary torsion dystonia. Mov Disord. 2000;15:954–9.
Stamelou M, Charlesworth G, Cordivari C, et al. The phenotypic spectrum of DYT24 due to ANO3 mutations. Mov Disord. 2014. doi:10.1002/mds.25802.
Vermeer S, Hoischen A, Meijer RP, et al. Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am J Hum Genet. 2010;87:813–9.
Huang F, Wang X, Ostertag EM, et al. TMEM16C facilitates Na+-activated K+ currents in rat sensory neurons and regulates pain processing. Nat Neurosci. 2013;16:1284–90.
Zech M, Gross N, Jochim A, et al. Rare sequence variants in ANO3 and GNAL in a primary torsion dystonia series and controls. Mov Disord. 2014;29:143–7.
Hopfner F, Bungeroth M, Pendziwiat M, et al. Rare variants in ANO3 are not a susceptibility factor in essential tremor. Parkinsonism Relat Disord. 2014;20:134–5.
Postma AG, Verschuuren-Bemelmans CC, Kok K, et al. Characteristics of dystonia in the 18p deletion syndrome, including a new case. Clin Neurol Neurosurg. 2009;111:880–2.
Esposito F, Addor MC, Humm AM, et al. GNAL deletion as a probable cause of dystonia in a patient with the 18p- syndrome. Parkinsonism Relat Disord. 2014;20:351–2.
Leube B, Hendgen T, Kessler KR, et al. Evidence for DYT7 being a common cause of cervical dystonia (torticollis) in central Europe. Am J Med Genet. 1997;74:529–32.
Han F, Racacho L, Lang AE, et al. Refinement of the DYT15 locus in myoclonus dystonia. Mov Disord. 2007;22:888–92.
Winter P, Kamm C, Biskup S, et al. DYT7 gene locus for cervical dystonia on chromosome 18p is questionable. Mov Disord. 2012;27:1819–21.
Kaur A. Rare autosomal dominant mutations in GNAL are associated with primary torsion dystonia. Clin Genet. 2013;84:211–2.
Miao J, Wan XH, Sun Y, et al. Mutation screening of GNAL gene in patients with primary dystonia from northeast China. Parkinsonism Relat Disord. 2013;19:910–2.
Charlesworth G, Bhatia KP, Wood NW. No pathogenic GNAL mutations in 192 sporadic and familial cases of cervical dystonia. Mov Disord. 2014;29:154–5.
Dufke C, Sturm M, Schroeder C, et al. Screening of mutations in GNAL in sporadic dystonia patients. Mov Disord. 2014. doi:10.1002/mds.25794.
Kumar KR, Lohmann K, Masuho I, et al. Mutations in GNAL: a novel cause of craniocervical dystonia. JAMA Neurol. 2014;71:490–4.
Saunders-Pullman R, Fuchs T, San Luciano M, et al. Heterogeneity in primary dystonia: lessons from THAP1, GNAL, and TOR1A in Amish-Mennonites. Mov Disord. 2014;29:812–8.
Xiao J, Zhao Y, Bastian RW, et al. Novel THAP1 sequence variants in primary dystonia. Neurology. 2010;74:229–38.
Luo AH, Cannon EH, Wekesa KS, et al. Impaired olfactory behavior in mice deficient in the α subunit of Go. Brain Res. 2002;941:62–71.
Ledoux MS, Dauer WT, Warner TT. Emerging common molecular pathways for primary dystonia. Mov Disord. 2013;28:968–81.
Bertran-Gonzalez J, Hakansson K, Borgkvist A, et al. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology. 2009;34:1710–20.
Liokatis S, Stutzer A, Elsasser SJ, et al. Phosphorylation of histone H3 Ser10 establishes a hierarchy for subsequent intramolecular modification events. Nat Struct Mol Biol. 2012;19:819–23.
LeDoux MS, Brady KA. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord. 2003;18:60–9.
LeDoux MS, Lorden JF, Ervin JM. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 1993;120:302–10.
Neychev VK, Gross RE, Lehericy S, et al. The functional neuroanatomy of dystonia. Neurobiol Dis. 2011;42:185–201.
Makino S, Kaji R, Ando S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80:393–406.
Weber YG, Kamm C, Suls A, et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology. 2011;77:959–64.
De Carvalho P, Sweadner KJ, Penniston JT, et al. Mutations in the Na+/K+-ATPase α3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–75.
Acknowledgements
Our dystonia research has been supported by grants from the National Institutes of Health (K08NS001593, R01EY012232, R01NS048458, R01NS050185, R01NS069936, and U54NS065701), the Dystonia Medical Research Foundation, the Bachmann-Strauss Dystonia & Parkinson Foundation, Tyler’s Hope for a Dystonia Cure, and the University of Tennessee Health Science Center Neuroscience Institute.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Jianfeng Xiao and Satya R. Vemula declare that they have no conflict of interest.
Mark S. LeDoux has received consultancy fees from Teva and Lundbeck. He has also received grants from the National Institutes of Health, the Cure Huntington Disease Initiative, and Prana. He has also received honorarium payments from Teva, Lundbeck, and UCB Pharma, as well as royalty payments from Elsevier.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Genetics
Rights and permissions
About this article

Cite this article
Xiao, J., Vemula, S.R. & LeDoux, M.S. Recent Advances in the Genetics of Dystonia. Curr Neurol Neurosci Rep 14, 462 (2014). https://doi.org/10.1007/s11910-014-0462-8
Published:
DOI: https://doi.org/10.1007/s11910-014-0462-8