
Abstract
Cinnamomum bodinieri is a useful plant that could be improved by increasing its cultivation rate. To explore the optimal concentration of exogenously applied IBA for root formation and development in the C. bodinieri citral type, micro-shoots and the fresh tissues of their bases were subjected to morphological and transcriptome analyses, respectively. When plantedQuery on a rooting medium (1/2 MS) with 1.5 mg/L IBA for 25 days, the rooting rates and root numbers were significantly higher than those obtained with 0, 0.5, 1.0, or 2.0 mg/L IBA treatment. Transcriptome analyses conducted 0 day, 7 days, 11 days, and 14 days after planting in the optimal rooting medium demonstrated that the IBA modified the plant’s carbohydrate metabolism and auxin signaling pathway. The root-specific expressions of TIR1, GH3, and SAUR were up-expressed while AUX/IAA was repressed, which contributed to the formation of lateral root initiation sites and root cell elongation. At the same time, d-glucose synthesis increased and the accumulation of starch decreased as more carbohydrates were used to form and develop roots instead of being used for the rapid cell division required for shoot growth. This study identified the optimal rooting medium for the C. bodinieri citral type and determined the molecular mechanisms involved in the regulation of exogenously applied IBA during root formation and development at the transcriptional level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cinnamomum bodinieri is an excellent tree species for the construction of forests. It has multiple applications in biomedicines, food additives, spices, cosmetics, and antibacterial agents (Liu et al. 2019; Wu et al. 2019). Recently, C. bodinieri has been cultivated as an economically important tree for its medicinal and aromatic properties (Chen et al. 2017; Li et al. 2018). Selective breeding has produced C. bodinieri with special uses, such as for spice strains with characteristic flavors and aromas, and high-potency medicinal cultivars (Chen et al. 2018). The C. bodinieri citral type was so named for the large amount of citral in its root bark, stem bark, and leaves. Citral, 3,7-dimethyl-2,6-octadienal is a key component of several essential oils and natural antioxidant substances (Bouzenna et al. 2017). Citral is also widely used in aromatherapy, food, pesticides, and as a fungicide (Wuryatmo et al. 2003). These traits give the C. bodinieri citral type high economic value and have made it the focus of recent scientific research.
At present, artificial planting of C. bodinieri citral type is mainly achieved via stem cutting cultivation and tissue culture; however, these techniques result in low survival rates and slow growth, which restrict production. It would be useful to improve the plant’s survival rate and rooting velocity and rate. The formation of plant roots is accompanied by cell division and expansion. Phytohormones play important roles in the regulation of cell division and expansion. The phytohormone auxin, which is mainly distributed in young and growing tissues and organs, is critical for plant growth and orchestrates many developmental processes (Yoshihisa et al. 2012). Indole-3-acetic acid (IAA) is recognized as the key auxin in most plants and it can be obtained by β-oxidation of indole-3-butyric acid (IBA), a second endogenous auxin (Woodward and Bartel 2005). It has been proven that the treatment of IBA enhances root regeneration in stem cuttings of different species, such as the mung bean (Pan and Tian 1999), Moraiolo (Ali et al. 2009), Camellia sinensis (Rout 2006) and so on. It is worth noting that the physiological role of auxin is twofold: a low concentration promotes plant growth by promoting cell proliferation and expansion (Iqbal et al. 2016), while an excessive concentration retards development (Björkman 2004). The present study was carried out to determine the optimum concentration of IBA needed to maximize the rooting potential of stooled shoots (Lal et al. 2007; Jamro et al. 2018).
In this study, fresh tissues from the micro-shoot base were used for morphological analysis to identify the optimal concentration of IBA for the rooting of C. bodinieri citral type micro-shoots. Based on transcriptome analysis, auxin signaling was observed to be involved in the subtle control and regulation of the carbohydrate metabolism process. A hypothesis integrating the metabolism adjustment and signaling alteration processes is presented.
Results
Global transcriptome analysis of the micro-shoot base in C. bodinieri citral type
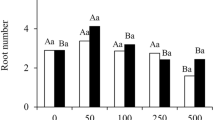
To identify the optimal concentration of exogenous auxins, different concentrations of IBA (0.5, 1.0, 1.5, 2.0 mg/L) were used to culture the micro-shoots of C. bodinieri citral type for 25 days. Significantly different effects were observed (Fig. 1A). The differences in root length and root thickness were not significant. However, it is worth noting that the rooting rates and root numbers first increased and then decreased with increases in IBA concentration. The rooting rate reached 75.00 ± 2.27% and the root number reached 3.50 ± 0.18 with 1.5 mg/L IBA treatment, which was significantly higher than that in other treatments. The next highest rates were observed in the 2.0 mg/L IBA treatment (68.75 ± 5.06% and 3.06 ± 0.33, respectively). These results demonstrate that the optimum rooting medium was 1/2 MS with 1.5 mg/L IBA.

Root development under the different concentration of IBA. A Rooting rates, root numbers, root length and diameter of 25 days C. bodinieri citral type micro-shoots after planting on the 1/2 MS media with 0.5, 1.0, 1.5 and 2.0 mg/L IBA. All the plants were grown under natural conditions and normal nutrients. B Phenotypes of 1 day, 3 days, 5 days, 7 days, 11 days and 14 days C. bodinieri citral type micro-shoots growing on the 1/2MS media with IBA(1.5 mg/L) (white bar = 1.5 cm). The 0 day, 7 days, 11 days and 14 days plants were chosen and named as ZH1 to ZH4 respectively. All bars represent means ± SD and those labeled with a letter are significantly different at P < 0.05 by Duncan’s test. Three biological replicates in the experiment were performed, and each biological replicate consists of 30 plants
Transcriptome analysis of the different stages of root development in the micro-shoot base of C. bodinieri citral type can provide a crucial systems-level insight into the molecular mechanisms underlying the formation and development of roots. When culturing on the 1/2 MS with 1.5 mg/L IBA, the morphology of the base was basically unchanged over the first 5 days. The base of the tissue culture seedlings expanded and calluses appeared at 7 days. Adventitious roots appeared in the tissue culture seedlings at 11 days and these grew and became obvious after culturing on the medium for 14 days. Based on these phenotypes, four time points representing major stages in root development were identified: 0 day, 7 days, 11 days, and 14 days. These were chosen for global transcriptome analysis and named ZH1, ZH2, ZH3, and ZH4, respectively (Fig. 1B).
Overall differentially expressed genes (DEGs) in the four developmental periods
To understand the molecular mechanism and regulation networks of C. bodinieri citral type during its various developmental periods, differentially expressed genes [log2 (relative expression level) ≥ 1 and ≤ − 1, padj ≤ 0.05, DEGs] in ZH2, ZH3 and ZH4 against with ZH1 were identified by RNA-Seq (Supplemental Tab. S1/S2/S3). Principal component analysis (PCA) of the DEGs revealed the separation of gene expression profiles among ZH1, ZH2 and ZH3 were quite different from each other. The distribution of the three biological replicates was aggregated, and the gene expression profiles of ZH4 were specifically similar with ZH3 (Fig. 2a). As summarized in Fig. 2b, compared with ZH1, 3307 upregulated and 3307 downregulated genes were observed in ZH2, 5037 upregulated and 4773 downregulated genes were observed in ZH3 compared with ZH1, and 4162 upregulated and 3655 downregulated genes were observed in ZH4 against ZH1. When comparing the DEGs of different groups, 1598 genes were consistently up-expressed and 1802 genes were down-expressed during the development periods, based on the Venn diagram in Fig. 2c. And there were more genes with the same expression pattern (1744 up-expressed, 1221 down-expressed) between the ZH4vsZH1 and ZH3vsZH1, that was just consistent with the aggregation of ZH3 and ZH4 in the Fig. 2a. The clustering analysis results of the relative expression levels of the overlapped genes in the four groups are also shown in Fig. 2d.

Overall differentially expressed genes (DEGs) during the development of C. bodinieri citral type. a Principal component analysis (PCA) of differentially expressed genes (log2 (relative expression level) ≥ 1 and ≤ -1, DEGs) in 4 groups. b The histogram of the statistical results for the DEGs in four groups. The blue for down-expressed genes and the gray for up-expressed genes. c Venn diagram of the overlap between the DEGs between ZH2 and ZH1, ZH3 and ZH1, ZH4 and ZH1. d The clustering analysis results of the relative expression levels of the different groups
Effects of IBA on cell division based on transcriptome analysis
Gene ontology terms for ZH2 vs ZH1, ZH3 vs ZH1 and ZH4 vs ZH1 showed different enrichment results for stress response, metabolism, biosynthesis, signaling, and protein transport, and so on. The up-expressed genes in ZH2 against ZH1 were enriched in the metabolism of carbohydrate and cellular glucan, while up-expressed genes in ZH4 against ZH1 were highly enriched in the plant response process. Genes regulating the division of plant cells were up-expressed during the whole rooting process (Fig. 3a, red dotted box). At the same time, all of the down-expressed genes in ZH2, ZH3, and ZH4 against ZH1 were enriched in the metabolic and biosynthetic processes of amide and peptide (Fig. 3a, dark blue dotted boxes). It was worth noticing that the upregulation of six TARGETING PROTEINs FOR XKLP2(TPX2), six CYCLINs and ten cellulose synthases indicates that the cell division was strong (Fig. 3b). Transcriptome analysis showed that the expression of genes involved in nucleosome assembly, transcript regulation, and cell division were significantly altered during root formation and development. Four genes encoding histone 4, four genes encoding histone 3.2, seven genes encoding histone 2, and two genes encoding histone 1 were significantly up-regulated, while two genes encoding deacetylase, which regulates the acetylation levels of histones and transcription factors, were down-expressed (Fig. 3c). The expression of genes was regulated by a complex interaction of cis-acting elements and trans-acting factors. There is no doubt that the TFs function effectively as trans-acting factors in plant growth, and the expressions of 6 bZIP, 3 ZF-HD, 2 HSP, 20 WRKY, 26 MYB TFs were improved significantly (Fig. 3c).

DEGs during the rooting were involved in nucleosome assembly, transcript regulation, cell division and growth. a GO enrichment analysis clustering results of the up-expressed (read) and down-expressed (blue) gene of ZH2, ZH3 and ZH4 in contrast to ZH1. b Up-regulated genes involved in cell division and growth. c Up-regulated genes involved in nucleosome assembly and transcription factor families. The absolute values of log2 (ZH2,3,4/ZH1) ≥ 1 and FDR < 0.001 were used as the criteria for DEGs. The color of the box represents up (red) and down (green)-regulated genes (ZH2,3,4/ZH1), and the value in the box is the log2 of the genes in the micro-shoots base
IBA changes the expression of the crucial auxin signaling pathway genes
To further compare the differences and similarities of these expression profiles, the DEGs in different lines were divided into seven groups according to the Venn diagram (group 7, overlapped in three lines; groups 4/5/6, overlapped between two lines; groups 1/2/3, DEGs in a single line; Fig. 4a). The GO classification of group 7 genes showed they were primarily involved in stimulus, especially the auxin response (Fig. 4a, black dotted box). Auxin plays a crucial role in the development of plants, especially the roots. By comparison, auxin was the most altered hormone signaling cascade, as demonstrated by the improved expressions of auxin response genes such as two TIR1 (transport inhibitor response 1-like protein, gene-CKAN_00789000, gene-CKAN_02338200), 3 GH3 (beta-glucosidase, gene-CKAN_00900200, gene-CKAN_02292900, gene-CKAN_00275400) and 7 SAUR-like auxin-responsive family genes, and by the downregulated expressions of 5 AUX/IAA (auxin-responsive protein, gene-CKAN_00961900, gene-CKAN_01790600, gene-CKAN_00406800, gene-CKAN_02372500, gene-CKAN_00662300). Meanwhile, the ARF (Arf GTPase activating protein) showed no significant change in expression level. Concomitantly, each two genes with significant changes in expression of TIR1, AUX/IAA, GH3 and SAUR families were selected for the real-time RT-PCR analysis, and details of the time-specific expression assay results shown in Fig. 4c were highly coincident with the RNA-seq result.

Go terms and pathway analysis of DEGs showed that auxin signaling process was significantly changed during the development. a Venn diagram (divided into seven groups) of the overlap and the GO enrichment analysis results of different groups. b DEGs involved in the auxin signaling pathway. The absolute values of log2 (ZH2,3,4/ZH1) ≥ 1 and FDR < 0.001 were used as the criteria for DEGs. The color of the box represents up (red) and down (green)-regulated genes (ZH2,3,4/ZH1), and the value in the box is the log2 (ZH2,3,4/ZH1) of the genes in the micro-shoots base. Schematic of the pathways modified according to the KEGG. c Expression profiles of genes (real-time RT-PCR results) involved in auxin signaling process during different stages of shoots rooting on the 1/2 MS with 1.5 mg/L IBA. All bars represent means ± sd (n = repeated 3 times) and those labeled with a letter are significantly different at P < 0.05 by Duncan’s test
Expression profiles of carbohydrate metabolism pathway genes
Based on the Venn assay, the GO classification of group 7 genes also indicated they were highly enriched in the carbohydrate metabolic process (Fig. 4a, red dotted box). A significant change was observed in the carbohydrate metabolism pathways, and the amylose synthesis pathway was decelerated (Fig. 5a). Four sucrose synthases (such as gene-CKAN_00708900 and gene-CKAN_02213000), three pfkB-like carbohydrate kinases (such as gene-CKAN_02194700, gene-CKAN_00641200), three trehalose phosphatases (such as gene-CKAN_01644300, gene-CKAN_01644300), seven β-glucosidases (such as gene-CKAN_00451800, gene-CKAN_01817400), three glycosyl hydrolases (such as gene-CKAN_00486900, gene-CKAN_02436500), seven glycosyl transferases (such as gene-CKAN_00696500, gene-CKAN_00786100) and three β-amylases, six phosphogluco mutases, and two hexokinases were significantly up-expressed in ZH2, ZH3, and ZH4 compared with ZH1 (Fig. 5a). At the same time, each two genes with significant changes in expression of sucrose synthases, pfkB-like carbohydrate kinases, trehalose phosphatases, β-glucosidase, glycosyl hydrolases and glycosyl transferases families were also selected for the real-time RT-PCR analysis to indicate the RNA-Seq assay results (Fig. 5b). All of these genes were up-expressed after the treatment of IBA. In addition, the range of expression levels were relatively large from ZH1 to ZH3. These results indicate that IBA might indirectly up-regulate the expression of amylase synthesis pathway genes. To explore the mechanism of root development from days 0 to 14 after planting with the 1/2 MS with 1.5 mg/L IBA, the starch and the soluble sugar contents of stems (part of the 1 cm to the end) and leaves in ZH1, ZH2, ZH3, and ZH4 were analyzed after 12 h light exposure (within the circadian rhythm of 12:12 h light:dark). The content of soluble sugars were increased, while the content of starch decreased with the time of culture (Fig. 5c).

Differential expression of representative genes involved in carbohydrate metabolism process. a DEGs in carbohydrate metabolism. The absolute values of log2 (ZH2,3,4/ZH1) ≥ 1 and FDR < 0.001 were used as the criteria for DEGs. The color of the box represents up (red) and down (green)-regulated genes (ZH2,3,4/ZH1), and the value in the box is the log2 (ZH2,3,4/ZH1) of the genes in the micro-shoots base. Schematic of the pathways modified according to the KEGG. b Expression profiles of genes involved in carbohydrate metabolism process during different stages of shoots rooting on the 1/2 MS with 1.5 mg/L IBA. c Staining results with Lugol’s solution. c Changes of starch and soluble sugar contents in ZH1, ZH2, ZH3 and ZH4. All bars represent means ± sd (n = repeated 3 times) and those labeled with a letter are significantly different at P < 0.05 by Duncan’s test
Discussion
Plant tissue cultivation always begin with the process of overcoming wounding response. Based on the RNA-Seq data from the 0 to 14 days after planting on the 1/2 MS with 1.5 mg/L IBA, the expressions of three wounding response protein (CKAN_01684900, CKAN_00960200, CKAN_01684700) and one wound-induced protein precursor (CKAN_01790800) were significantly induced, whose expression in ZH4 showed 2.08, 14.5, 1.5 and 1.48-fold of that in ZH1 respectively. This result indicates the accuracy of transcriptome sampling and the reliability of transcriptome data from a side.
The GO, KEGG pathway, and relative expression level clustering analyses demonstrate that during the formation and development of roots, DEGs were enriched in the auxin signaling pathway (Fig. 4b). Previous studies show that auxin plays a central role in regulating root growth and development by regulating the expressions of TIR1, AUX/IAA, ARF, GH3, and SAUR. TIR1 was the recognition component of the SCF complex associated with the ubiquitin–proteasome pathway involved in auxin signaling and the receptor for IAA (Walsh et al. 2006). Gene-CKAN_00789000 and gene-CKAN_02338200 are the orthologs of AUXIN F-BOX PROTEIN 5 (AFB5) and AtTIR1 in Arabidopsis, and are highly expressed in the root, hypocotyl, and cotyledon cells. AFB5 plays an important role in the development of lateral roots (Ishimaru et al. 2018), and the T-DNA insertion mutants afb2-3, afb5-5, and tir1-1 are resistant to IAA and the synthetic auxin picloram in dwarf plants (Prigge et al. 2016). Mutant tir1-1 is also resistant to auxin-mediated inhibition of root elongation (Ishimaru et al. 2018; Biswas et al. 2019). These results are consistent with the improvements in the expressions of gene-CKAN_00789000 and gene-CKAN_02338200 from ZH1 to ZH4 (Fig. 4c). The AtGH3.17 orthologs in C. bodinieri citral type are gene-CKAN_00900200 and gene-CKAN_00275400, which belong to the GH3 group II family and encode an IAA-amido synthase (Pierdonati et al. 2019). Prior study indicates that PIN5 and GH3.17 can regulate auxin transport and control auxin levels throughout the root meristem to regulate its size and root growth (Di Mambro et al. 2019). The up-regulated gene-CKAN_00900200 and gene-CKAN_00275400 were positively correlated with the development trend of the roots. Gene-CKAN_00358400/gene-CKAN_01922100, which is the ortholog of SAUR41 (SMALL AUXIN UPREGULATED 41) in C. bodinieri citral type, encodes a clade III SAUR gene with a distinctive expression pattern in root meristems and is also involved in auxin transport and the formation of a radial root pattern (Qiu et al. 2013, 2019).
The AUX/IAA proteins are short-lived nuclear proteins sharing four conserved domains (Park et al. 2002). In this study, gene-CKAN_01790600, gene-CKAN_00406800 and three other genes, the orthologs of AtIAA2/3 (INDOLE-3-ACETIC ACID INDUCIBLE 2/3) in Arabidopsis (which can regulate lateral root development by activating the expression of LBD16/18; Lee et al., 2015), were repressed in ZH2, ZH3, and ZH4 against ZH1. The shy2-101 gene, a mutant of IAA3, had more lateral root initiation sites in the distal root region but the subsequent periclinal cell division was inhibited (Goh et al. 2012), which explains the root formation and retarded plant growth. In summary, analysis of the DEGs in the auxin signaling pathways revealed that these IAA-modulated developmental processes were altered in the IBA treatments. They improved the formation and development of lateral roots but induced growth retardation.
AtSUS5 is the ortholog of gene-CKAN_00708900/gene-CKAN_02213000 in Arabidopsis, and encodes the crucial SUCROSE SYNTHASE, which catalyzes the reversible conversion of sucrose and a nucleoside diphosphate into a corresponding nucleoside diphosphate-glucose and fructose (Bieniawska et al. 2007). Research in recent decades has shown that SUS is not a determining factor in the biosynthesis of cellulose and starch, but has been confirmed to be involved in the sucrose-starch and sucrose–cellulose conversion processes (Baroja-Fernández et al. 2012). These results indicate that there is an active carbohydrate conversion process occurring during root formation. Gene-CKAN_02194700/gene-CKAN_00641200 encodes the PfkB family carbohydrate kinase with fructokinase activity and is essential for the development and growth of plants (Gilkerson et al. 2012). Trehalose phosphatase (TPS), which encodes an enzyme putatively involved in trehalose biosynthesis, is also a key rate-limiting enzyme in the conversion of reducing sugar to non-reducing sugar, and then to direct energy (glucose; Eastmond et al. 2002). TPS can regulate starch and sucrose degradation and is involved in the regulation of glucose sensing and signaling genes during plant development (Schluepmann et al. 2003; Gómez et al. 2006). β-Glucosidase (BGLU), such as gene-CKAN_00451800 or gene-CKAN_01817400, is also exclusively expressed in roots (Nitz et al. 2001; Ahn et al. 2010) and is the crucial enzyme for the synthesis of d-glucose. It is worth noting that SUS, TPS, and BGLU, which took part in sucrose utilization, were all up-expressed during the formation and development of roots (Fig. 5a, b).
Amylose is another major source of glucose. ATGATL1 and IRX9, the orthologs of gene-CKAN_00696500 and gene-CKAN_00786100 in Arabidopsis, encode glycosyl transferase, which contributes to the utilization of starch glycogen and the biosynthesis of glucuronoxylan and d-glucose (Lao et al. 2003; Shao et al. 2004; Lee et al. 2007). Starch glycogen can also degrade to d-glucose through maltose based on β-amylase, such as BAM1/3, which regulates cell division and differentiation to organize cell layers by regulating CLV1-dependent CLV3-mediated signaling in meristem maintenance (DeYoung et al. 2006; DeYoung and Clark 2008). The overexpression of glycosyl transferases and β-amylases was identified by RT-PCR (Fig. 5b), and the leaves and stems of ZH2, ZH3, and ZH4 undoubtedly had reduced accumulations of starch compared to that of ZH1 (Fig. 5c).
All these results indicate that exogenously applied IBA modifies carbohydrate metabolism and the auxin signaling pathway (Fig. 6). On the one hand, the root-expressed genes TIR1, GH3, SAUR, and AUX/IAA improved the formation of lateral root initiation sites and root cell elongation. On the other hand, d-glucose synthesis was increased and disaccharide (sucrose) and starch utilization was accelerated, potentially due to the kernels attempting to use more carbohydrates to form and develop roots instead of undergoing rapid cell division and growing shoots.

The proposed model for the regulation of roots development by exogenous IBA. The root expressed TIR1, GH3, SAUR and AUX/IAA improved the formation of lateral root initiation sites, the root cell elongation. d-Glucose synthesis was increased, disaccharide (sucrose) and starch utilization was accelerated
Materials and methods
Plant materials
Plants were grown on 1/2 MS agar plates at 25 °C under natural daylight conditions (12:12 h light:dark cycle, with a 4000 lx incandescent lamp as a supplementary light source). For RNA extraction and content measurement, fresh tissues of the micro-shoots base (10 mm, 8 plants per sample) taken 0 day, 7 days, 11 days, and 14 days after planting on the 1/2 MS media were collected. External contaminants were removed by washing with ultrapure water three times, then samples were frozen in liquid nitrogen and stored at − 80 °C.
Transcriptome analysis
All samples were sent to Novogene (Beijing, China) for extraction, sequencing, and bioinformatics analyses. mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First strand synthesis reaction buffer (5 ×). First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′ ends of the DNA fragments, NEBNext Adaptor with a hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments that were preferentially 250–300 bp in length, the library fragments were purified with an AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μL USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min, followed by 5 min at 95 °C before PCR analysis. Then, PCR was performed with Phusion High-fidelity DNA polymerase, Universal PCR primers and index (X) Primer. Finally, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system.
Clustering and sequencing
Clustering of the index-coded samples was performed on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform and 150 bp paired-end reads were generated.
Differential expression analysis
Differential expression analysis of four groups (three biological replicates per condition) was performed using the DESeq2 R package (1.16.1). DESeq2 provides statistical routines for determining differential expressions in digital gene expression data using a model based on the negative binomial distribution. The resulting P values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P value < 0.05 found by DESeq2 were assigned as differentially expressed. The selection of differentially expressed genes was based on FDR ≤ 0.001 and an absolute value of Log2 (relative expression level, compared with WT) ≥ 1 and ≤ − 1.
GO and KEGG enrichment analysis of differentially expressed genes
Case special deep analysis based on DEGs, including GO and KEGG pathway enrichment analysis, was done by our group. Gene ontology (GO) enrichment analysis of differentially expressed genes was implemented by the cluster Profiler R package, in which gene length bias was corrected. GO terms with corrected P values < 0.05 were considered significantly enriched by differential expressed genes. KEGG is a database for understanding the high-level functions and utilities of biological systems, such as cells, organisms, and ecosystems, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (https://www.genome.jp/kegg/). We used the cluster Profiler R package to test the statistical enrichment of differentially expressed genes in KEGG pathways.
Plant RNA extraction and real-time RT-PCR
The total RNA was extracted using Trizol reagent (Takara, Japan). An amount of 500 ng total RNA was used for inverse transcription with a Transcriptor First Strand cDNA Synthesis Kit (Takara, Japan). We diluted the cDNA ten times and then used it as the template for real-time PCR (RT-PCR). The RT-PCR reaction system comprised SYBR® Premix Ex TaqTM (2 ×) 5 μL, PCR Forward Primer (10 μM) 0.2 μL, PCR Reverse Primer (10 μM) 0.2 μL, cDNA template 1 μL, and distilled deionized water up to 10 μL. The PCR process used the following conditions: 95 °C for 15 s, 56 °C for 30 s, and 72 °C for 30 s for 40 cycles.
Determination of soluble sugars and starch contents
Starch and soluble sugars were determined with Anthrone reagent (Hansen and Moller 1975; Taylor et al. 1982) with the leaves and the stems (1 cm to the end) as the materials (three biological replications, five plants for each biological replication).
References
Ahn YO, Shimizu BI, Sakata K et al (2010) Scopolin-hydrolyzing β-glucosidases in roots of Arabidopsis. Plant Cell Physiol 51:132–143. https://doi.org/10.1093/pcp/pcp174
Ali A, Ahmad T, Abbasi NA, Hafiz IA (2009) Effect of different concentrations of auxins on In vitro rooting of olive cultivar “Moraiolo”. Pak J Bot 41:1223–1231
Baroja-Fernández E, Muñoz FJ, Li J et al (2012) Sucrose synthase activity in the sus1/sus2/sus3/sus4 Arabidopsis mutant is sufficient to support normal cellulose and starch production. Proc Natl Acad Sci 109:321 LP–326 LP. https://doi.org/10.1073/pnas.1117099109
Bieniawska Z, Paul Barratt DH, Garlick AP et al (2007) Analysis of the sucrose synthase gene family in Arabidopsis. Plant J 49:810–828. https://doi.org/10.1111/j.1365-313X.2006.03011.x
Biswas MS, Fukaki H, Mori IC et al (2019) Reactive oxygen species and reactive carbonyl species constitute a feed-forward loop in auxin signaling for lateral root formation. Plant J. https://doi.org/10.1111/tpj.14456
Björkman T (2004) Effect of Trichoderma colonization on auxin-mediated regulation of root elongation. Plant Growth Regul 43:89–92. https://doi.org/10.1023/B:GROW.0000038260.85276.82
Bouzenna H, Hfaiedh N, Giroux-Metges MA et al (2017) Biological properties of citral and its potential protective effects against cytotoxicity caused by aspirin in the IEC-6 cells. Biomed Pharmacother 87:653–660. https://doi.org/10.1016/j.biopha.2016.12.104
Chen C, Zheng Y, Liu S et al (2017) The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ. https://doi.org/10.7717/peerj.3820
Chen C, Zheng Y, Zhong Y et al (2018) Transcriptome analysis and identification of genes related to terpenoid biosynthesis in Cinnamomum camphora. BMC Genom. https://doi.org/10.1186/s12864-018-4941-1
DeYoung BJ, Bickle KL, Schrage KJ et al (2006) The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase-like proteins are required for meristem function in Arabidopsis. Plant J 45:1–16. https://doi.org/10.1111/j.1365-313X.2005.02592.x
DeYoung BJ, Clark SE (2008) BAM receptors regulate stem cell specification and organ development through complex interactions with CLAVATA signaling. Genetics 180:895–904. https://doi.org/10.1534/genetics.108.091108
Di Mambro R, Svolacchia N, Dello Ioio R et al (2019) The lateral root cap acts as an auxin sink that controls meristem size. Curr Biol 29:1199–1205.e4. https://doi.org/10.1016/j.cub.2019.02.022
Eastmond PJ, Van Dijken AJH, Spielman M et al (2002) Trehalose-6-phosphate synthase 1, which catalyses the first step in trehalose synthesis, is essential for Arabidopsis embryo maturation. Plant J 29:225–235. https://doi.org/10.1046/j.1365-313x.2002.01220.x
Gilkerson J, Perez-Ruiz J, Chory J, Callis J (2012) The plastid-localized pfkB-type carbohydrate kinases FRUCTOKINASE-LIKE 1 and 2 are essential for growth and development of Arabidopsis thaliana. BMC Plant Biol 12:102
Goh T, Kasahara H, Mimura T et al (2012) Multiple AUX/IAA-ARF modules regulate lateral root formation: the role of Arabidopsis SHY2/IAA3-mediated auxin signalling. Philos Trans R Soc B Biol Sci 367:1461–1468. https://doi.org/10.1098/rstb.2011.0232
Gómez LD, Baud S, Gilday A et al (2006) Delayed embryo development in the ARABIDOPSIS TREHALOSE-6-PHOSPHATE SYNTHASE 1 mutant is associated with altered cell wall structure, decreased cell division and starch accumulation. Plant J 46:69–84. https://doi.org/10.1111/j.1365-313X.2006.02662.x
Hansen J, Moller IB (1975) Analysis of starch and soluble sugars with anthrone reagent. Anal Biochem 68:87–94
Iqbal MA, Khalid M, Zahir ZA, Ahmad R (2016) Auxin producing plant growth promoting rhizobacteria improve growth, physiology and yield of maize under saline field conditions. Int J Agric Biol 18:37–45. https://doi.org/10.17957/IJAB/15.0059
Ishimaru Y, Hayashi K, Suzuki T et al (2018) Jasmonic acid inhibits auxin-induced lateral rooting independently of the CORONATINE INSENSITIVE1 receptor. Plant Physiol 177:1704–1716. https://doi.org/10.1104/pp.18.00357
Jamro MMUR, Shah AN, Nizamani FK (2018) Effects of IBA and NAA on integrated root development in aerial offshoots of Phoenix dactylifera L. Bangladesh J Bot 47:287–292
Lal S, Tiwari JP, Awasthi P, Singh G (2007) Effect of IBA and NAA on rooting potential of stooled shoots of guava (Psidium guajava L.) cv. sardar. Acta Hortic 735:193–196
Lao NT, Long D, Kiang S et al (2003) Mutation of a family 8 glycosyltransferase gene alters cell wall carbohydrate composition and causes a humidity-sensitive semi-sterile dwarf phenotype in Arabidopsis. Plant Mol Biol 53:647–661. https://doi.org/10.1023/b:plan.0000019074.60542.6c
Lee C, Zhong R, Richardson EA et al (2007) The PARVUS gene is expressed in cells undergoing secondary wall thickening and is essential for glucuronoxylan biosynthesis. Plant Cell Physiol 48:1659–1672. https://doi.org/10.1093/pcp/pcm155
Lee HW, Cho C, Kim J (2015) LBD16 and LBD18 act downstream of the AUX1 and LAX3 auxin influx carriers to control lateral root development in Arabidopsis thaliana. Plant Physiol 168:15.00578. https://doi.org/10.1104/pp.15.00578
Li YR, Fu CS, Yang WJ et al (2018) Investigation of constituents from Cinnamomum camphora (L.) J. Presl and evaluation of their anti-inflammatory properties in lipopolysaccharide-stimulated RAW 264.7 macrophages. J Ethnopharmacol 221:37–47. https://doi.org/10.1016/j.jep.2018.04.017
Liu X, Meng Y, Zhang Z et al (2019) Functional nano-catalyzed pyrolyzates from branch of Cinnamomum camphora. Saudi J Biol Sci. https://doi.org/10.1016/j.sjbs.2019.06.003
Nitz I, Berkefeld H, Puzio PS, Grundler FMW (2001) Pyk10, a seedling and root specific gene and promoter from Arabidopsis thaliana. Plant Sci 161:337–346. https://doi.org/10.1016/S0168-9452(01)00412-5
Pan R, Tian X (1999) Comparative effect of IBA, BSAA and 5,6-Cl2-IAA-Me on the rooting of hypocotyl in mung bean. Plant Growth Regul. https://doi.org/10.1023/A:1006154426941
Park JY, Kim HJ, Kim J (2002) Mutation in domain II of IAA1 confers diverse auxin-related phenotypes and represses auxin-activated expression of Aux/IAA genes in steroid regulator-inducible system. Plant J 32:669–683. https://doi.org/10.1046/j.1365-313X.2002.01459.x
Pierdonati E, Unterholzner SJ, Salvi E et al (2019) Cytokinin-dependent control of GH3 group II family genes in the arabidopsis root. Plants. https://doi.org/10.3390/plants8040094
Prigge MJ, Greenham K, Zhang Y et al (2016) The Arabidopsis auxin receptor F-box proteins AFB4 and AFB5 are required for response to the synthetic auxin picloram. G3 Genes Genomes Genet 6:1383–1390. https://doi.org/10.1534/g3.115.025585
Qiu T, Chen Y, Li M et al (2013) The tissue-specific and developmentally regulated expression patterns of the SAUR41 subfamily of SMALL AUXIN UP RNA genes: potential implications. Plant Signal Behav. https://doi.org/10.4161/psb.25283
Qiu T, Qi M, Ding X et al (2019) The SAUR41 subfamily of SMALL AUXIN UP RNA genes is abscisic acid-inducible to modulate cell expansion and salt tolerance in Arabidopsis thaliana seedlings. Ann Bot. https://doi.org/10.1093/aob/mcz160
Rout GR (2006) Effect of auxins on adventitious root development from single node cuttings of Camellia sinensis (L.) Kuntze and associated biochemical changes. Plant Growth Regul. https://doi.org/10.1007/s10725-005-5665-1
Schluepmann H, Pellny T, Van Dijken A et al (2003) Trehalose 6-phosphate is indispensable for carbohydrate utilization and growth in Arabidopsis thaliana. Proc Natl Acad Sci USA 100:6849–6854. https://doi.org/10.1073/pnas.1132018100
Shao M, Zheng H, Hu Y et al (2004) The gaolaozhuangren1 gene encodes a putative glycosyltransferase that is critical for normal development and carbohydrate metabolism. Plant Cell Physiol 45:1453–1460. https://doi.org/10.1093/pcp/pch168
Taylor JS, Blake TJ, Pharis RP (1982) The role of plant hormones and carbohydrates in the growth and survival of coppiced Eucalyptus seedlings. Physiol Plant. https://doi.org/10.1111/j.1399-3054.1982.tb04522.x
Walsh TA, Neal R, Merlo AO et al (2006) Mutations in an auxin receptor homolog AFB5 and in SGT1b confer resistance to synthetic picolinate auxins and not to 2,4-dichlorophenoxyacetic acid or indole-3-acetic acid in Arabidopsis. Plant Physiol 142:542–552. https://doi.org/10.1104/pp.106.085969
Woodward AW, Bartel B (2005) Auxin: regulation, action, and interaction. Ann Bot 95:707–735
Wu K, Lin Y, Chai X et al (2019) Mechanisms of vapor-phase antibacterial action of essential oil from Cinnamomum camphora var. linaloofera Fujita against Escherichia coli. Food Sci Nutr. https://doi.org/10.1002/fsn3.1104
Wuryatmo E, Klieber A, Scott ES (2003) Inhibition of citrus postharvest pathogens by vapor of citral and related compounds in culture. J Agric Food Chem 51:2637–2640. https://doi.org/10.1021/jf026183l
Yoshihisa K, Kazuyoshi T, Masafumi H et al (2012) Arabidopsis ABCB21 is a facultative auxin importer/exporter regulated by cytoplasmic auxin concentration. Plant Cell Physiol 53(12):2090–2100
Acknowledgements
This study was supported by the Natural Science Foundation of China (31600497), the Science and Technology Department of Jiangxi Education, China (GJJ151114), the Key Research and Development project of Science and Technology Department of Jiangxi province, China (20171ACH80016, 20192BBFL60012).
Author information
Authors and Affiliations
Contributions
ZJ designed the project. ZX analyzed data and wrote the paper. ZX performed experiments. ZX contributed samples, materials, or data. BZ and FL helped with the data analysis and examined the results.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, Z., Jin, Z., Zhang, B. et al. Effects of IBA on rooting ability of Cinnamomum bodinieri citral type micro-shoots from transcriptomics analysis. Plant Biotechnol Rep 14, 467–477 (2020). https://doi.org/10.1007/s11816-020-00626-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-020-00626-5