
Abstract
Type-I hot corrosion behavior of CM247LC superalloy is evaluated in the air at 950 °C against low (3 to 4), intermediate (7 to 9), and high (12 to 14 mg cm−2) Na2SO4 deposits. Duration of thermal exposure is varied from a very short duration of 5 minutes to long duration of 1000 hours. The alloy shows poor corrosion resistance and undergoes complete disintegration after 500 hours of thermal exposure. Degradation of alloy increases with the increase in the duration of exposure as well as the initially deposited amount of the salt. Based on the systematic analysis of the corrosion scale, degradation mechanisms supported by the microstructural evidence are proposed. Fluxing, sulfidation-oxidation, and sulfide-undercutting are reported as the primary degradation mechanisms for CM247LC alloy in the presence of Na2SO4. Self-sustaining degradation of alloy leading to complete disintegration of specimen is caused by the changeover of fluxing mechanism from basic fluxing to alloy-induced fluxing.
Similar content being viewed by others

1 Introduction
The Ni-base superalloy components such as blades and vanes operating in the turbine engines undergo hot corrosion damage. Material degradation by hot corrosion occurs due to the reaction of a superficial film of salt with the metallic components at high temperatures. The corrosive salts such as sulfates, chlorides, and vanadates are formed in situ during the operation of the engine.[1,2,3,4] It is well established that hot corrosion causes greater damage to the material than pure high-temperature oxidation.[5,6,7,8,9,10,11,12] Fluxing and sulfidation-oxidation are two well-accepted mechanisms for hot corrosion of Ni-base superalloys.[7,8,13,14,15,16,17,18,19,20] The corrosion mechanism is affected by the service conditions as well as the elements contained in the superalloy.[1,8,10,15,16] For example, the degradation of superalloys at temperatures above the melting point of salt occurs by basic fluxing.[1,19,20] On the other hand, acidic fluxing occurs at temperatures lower than the melting point of salt.[1,19] The oxides of the refractory elements such as W, Mo, and V present in the alloy are known to promote acidic fluxing even at temperatures above the melting point of salt.[1,19,20] Considering the presence of multifarious alloying elements in the advanced superalloys, their hot corrosion behaviors can be expected to be complex and cannot be stereotyped.
The present study examines the Type-I hot corrosion behavior of the directionally solidified (DS) Ni-base superalloy CM247LC. This alloy is optimized for casting of turbine blades and vanes operating in the hot sections of advanced gas turbine engines and has a maximum temperature capability of 1000 °C.[21,22,23,24,25] Reports on the hot corrosion behavior of the above alloy are limited in the open literature.[26,27,28] Gurrappa examined the comparative corrosion behavior of CM247LC immersed in pure Na2SO4 and Na2SO4-NaCl mixture over a temperature range from 700 °C to 1000 °C.[26] The study proposed that the formation of superficial Al2O3 layer prevented severe hot corrosion during the initial 25 hours of immersion in pure Na2SO4 molten salt. The presence of NaCl in molten Na2SO4, however, induced cracking and spallation of the protective Al2O3 scale leading to accelerated degradation of the alloy by acidic fluxing. Sumner et al. examined the hot corrosion behavior of CM247 alloy under dynamic flow conditions during exposure to combustion flame in a burner rig test and reported a short incubation stage preceding rapid corrosion.[27] Tsao et al. compared the hot corrosion behavior of this alloy with that of high entropy superalloys in the presence of Na2SO4-25 pct NaCl deposit using immersion and salt film techniques.[28] It was reported that degradation of the alloy ceased during the 20 hours of exposure in the salt film test due to the depletion of salt with time and only oxidation occurred in the latter part of the 20 hours exposure. The degradation continued only after washing and re-coating of the specimen was done after 20 hours of exposure. However, aggravated degradation occurred in the immersed samples due to the abundance of molten salt.
The objective of the present study is to evaluate the progressive corrosion damage in the directionally solidified (DS) CM247LC superalloy caused by Na2SO4 salt during isothermal exposure at 950 °C in air for various durations ranging between 5 minutes and 1000 hours. The gradual microstructural evolution of the scale during corrosion is characterized and the operating mechanisms for corrosion ascertained. The study reports synergistic effects of fluxing and sulfidation-oxidation mechanisms. It proposes sulfide-undercutting at the corrosion front and examines its role in the degradation process.
2 Experimental Details
DS CM247LC Ni-base superalloy, having a nominal composition of (in wt pct) Ni 62.7-Co 9.2-W 8.5-Cr 8.1-Al 5.6-Ta 3.2-Hf 1.3-Ti 0.7-Mo 0.5-C 0.05-Zr 0.015-B 0.015, was obtained in the form of 12 mm diameter and 100 mm long rods. The [001] growth direction of the grains was along the length of the rods. Cylindrical specimens having 4 mm diameter and 25 mm length were cut from the rods using wire electro-discharge machining (EDM). The above specimen dimensions conformed to the BS ISO21608:2012 standard recommended for isothermal hot corrosion tests.[29] The samples were grit blasted to remove the oxide and recast layer formed on the surface during the EDM process and ultrasonically cleaned in the acetone bath. Subsequently, each sample was heated to 180 °C in an oven and a film of Na2SO4 was deposited on the sample by spraying a saturated aqueous solution of Na2SO4 salt. The specimens were weighed before and after the deposition of the salt layer to determine the weight of the deposited salt. The specific weight change, i.e., weight of the deposited salt divided by the initial surface area of the sample, was measured. Samples with three different salt deposits, namely, 3 to 4, 7 to 9, and 12 to 14 mg cm-2, were prepared for assessing the effect of initially available salt concentration on the hot corrosion behavior of the alloy. These samples are referred to as 3MC, 7MC, and 12MC, respectively, in the subsequent text.
The melting temperature of the Na2SO4 salt used in this study was determined as 883.5 °C using a Setaram Labsys Evo Differential Scanning Calorimeter (DSC). The salt-coated specimens were subjected to isothermal exposure at 950 °C in air using a muffle furnace for various durations of 10, 20, 40, 60, 80, 100, 150, 200, 500, and 1000 hours. The tests were carried out in Type-I hot corrosion regime[8,9,10] by selecting the test temperature above the melting temperature of salt and ensuring the formation of molten Na2SO4 salt film on the surface of the samples. A corresponding set of bare superalloy specimens, i.e., without any salt coating, was oxidized separately to the similar isothermal conditions at 950 deg for comparison purpose. These bare samples which were oxidized without any surface layer of salt are referred to as 0MC in the text. The specific change in weight caused by hot corrosion/oxidation exposure was measured for all the specimens and thermo-gravimetry plots generated with respect to the duration of thermal exposure. In order to probe the evolution of the corrosion products during the initial stages of hot corrosion, a few 3MC specimens were also exposed for short durations between 5 minutes and 1 hour at 950 °C in air.
The corroded specimens were encapsulated in resin using vacuum impregnated cold mounting process. The cross-sections of the encapsulated specimens were mounted and polished for microstructural characterization. Encapsulation of the corroded specimens prior to mounting and polishing prevented damage to the corrosion scale during mounting and polishing. Standard metallographic techniques were used for polishing the mounts. Precautionary measures, such as gentle dry paper polishing, final diamond polishing using oil-based diamond suspension and cleaning of the mounts using methanol, were adopted while polishing and cleaning of mounts to avoid dissolution of the corrosion product. The mounts were observed under an optical microscope (Leitz Metalloplan) and FEI Quanta 400D scanning electron microscope (SEM) operating at 20 kV coupled with an energy dispersive spectroscopy (EDS) facility. Elemental analysis of the corrosion scale was carried out using a Cameca SX-100 electron probe microanalyzer (EPMA) operating at 20 kV.
3 Results
3.1 Visual Inspection
The salt layer deposited on the samples prior to the hot corrosion test was well adherent and uniform in thickness, which is evident from the cross-section microstructure and the bright appearance of the sample surfaces (Figure 1(a)). After the corrosion tests for various durations, the samples appear greenish-blue and exhibit the formation of a loose scale having many cracks on the surface, as typically shown for the 3MC sample (Figures 1(b) through (d)). The thickness of corrosion scale and the extent of its cracking increase with the increasing duration of exposure. In fact, the samples undergo complete corrosion-induced disintegration after long durations of hot corrosion exposure such as after 1000 hours (Figure 1(d)).

Photographs for the 3MC samples after exposure in air at 950 °C for various durations: (a) 0 h, i.e., as-salt-coated without exposure, (b) 40 h, (c) 80 h, and (d) 1000 h. The uniform as-deposited salt layer in the sample prior to thermal exposure is evident from the cross-sectional micrograph shown in the inset in (a)
3.2 Weight Change Behavior
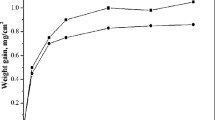
The weight change plots for the samples applied with various thicknesses of salt deposit, i.e., 3MC, 7MC, and 12MC, corresponding to exposure at 950 °C are shown in Figure 2. The weight change plot for the 0MC specimen, which did not have any salt deposit and its weight change is caused by oxidation alone and not by salt corrosion, is also included in Figure 2 for comparison. It is evident from the weight change plots that the weight gain and degradation caused by hot corrosion is significantly higher than that caused by oxidation alone. The salt-coated specimens show about 200 times higher specific weight gain than 0MC after 1000 hours of exposure. All the salt-coated samples register a rapid increase in weight during 500 hours of thermal exposure and the extent of weight gain increases with the increase in salt content. For instance, the weight gain after 500 hours of exposure is higher at ~ 250 mg cm−2 for the 12MC sample than that of the 7MC (230 mg cm−2) and 3MC samples (~ 210 mg cm−2). The samples do not exhibit an appreciable change in weight beyond 500 hours and the weight change values of the samples after 1000 hours of exposure are similar to those after 500 hours for all the salt deposited samples (Figure 2), which indicates that most of the hot corrosion damage of the alloy occurs during the initial 500 hours in all the salt deposited samples. It is evident from the weight change plot (Figure 2) and the sample photograph shown in Figure 1(d) that the salt content as low as 3 to 4 mg cm−2 (as in the 3MC sample) is sufficient to cause heavy corrosion damage and degradation of the superalloy during long thermal exposure.

Specific weight change plots for various samples at 950 °C. Inset shows the plot corresponding to oxidation of the specimen sans salt layer, i.e., 0MC. The axis labels in the inset are the same as that of the main figure
The weight change behavior of the 3MC sample during the initial stages of exposure, i.e., for short durations ranging between 5 minutes and 1 hours at 950 °C in air, is shown in Figure 3. The weight gain is significantly low at 0.3 to 0.5 mg cm−2 up to 30 minutes. of exposure. Subsequently, it increases rapidly to 6 mg cm−2 after 60 minutes (Figure 3). The low weight change during the initial 30 minutes indicates that this period constitutes the initiation stage (or incubation stage) of hot corrosion. The rapid weight gain beyond this duration corresponds to the propagation stage of hot corrosion (Figure 3). The transition from initiation stage to propagation stage is also supported by the microstructural observations elaborated in Section III–C–3.

Plot showing the variation of specific weight gain versus exposure time for 3MC sample. The initiation and propagation stages of hot corrosion are indicated
The extent of degradation by hot corrosion can be quantified in terms of the unaffected cross-sectional area of a specimen remaining after thermal exposure. Therefore, a parameter termed as ‘UCA’(in pct) is introduced in the present study. The UCA represents the percentage of the average unaffected cross-section, calculated using the average diameter (Davg) of the unaffected area, with respect to the initial cross-section of the specimen. Following the standard ISO 26146:2012(E),[30] Davg which represents the remaining metal is measured by observing the polished radial cross-sections of cylindrical corroded samples. The scheme for the measurement of UCA is indicated in Figure 4(a). D0 mentioned in the schematic (Figure 4(a)) is the original diameter of the specimen before test. The comparison between the UCA values for the samples containing lower (3MC) and higher (12MC) amounts of the initially deposited salt is shown in Figure 4(b). It is evident that irrespective of the initial salt content, the UCA decreases with the increase in the duration of hot corrosion exposure. For example, the UCA for 3MC and 12MC samples decreases from 95 to 14 and 94 to 6 pct, respectively, with the increase in exposure duration from 10 to 500 hours (Figure 4(b)). Though the short-term corrosion behavior (until 80 hours) appears similar, the extent of the decrease in UCA is higher for the samples applied with thicker salt deposits. The UCA is reduced to half of its original value after ~ 350 hours exposure for 3MC and ~ 200 hours exposure for 12MC. This implies that thick salt deposits attack substrate more severely than thin salt deposits. The UCA value decreases to about zero during 500 to 1000 hours of exposure for both the 3MC and 12 MC samples (Figure 4(b)), which indicates complete disintegration of the specimen as evidenced from Figure 1(d). The fact that the initially deposited salt layer was not replenished in the present study demonstrates the self-sustaining nature of hot corrosion of CM247 alloy under the molten salt film.

(a) Schematic showing scheme for the measurement of Unaffected Cross-sectional Area (UCA) and (b) UCA vs time plot for 3MC and 12MC specimens. Total disintegration of the specimens for both the conditions was observed at 1000 h of exposure
3.3 Microstructural Degradation After Thermal Exposure
3.3.1 Samples without salt deposit
The typical cross-sectional microstructure of 0MC sample after 1000 hours of exposure at 950 °C is shown in Figure 5. The formation of a thin layer of oxide scale on the surface of the oxidized specimen is evident. The elemental maps shown in Figure 5 indicate that the oxide scale is constituted of oxides of Ni, Al, and Cr. The composition of the oxide scale is similar to that reported by Das et al.[31] Oxidation in sub-surface regions of the superalloy is also observed (Figure 5). The oxidation behavior and oxidation-induced degradation mechanisms for the CM247LC alloy have been reported in our earlier publication.[31]

Elemental maps for oxidized alloy (0MC) exposed for 1000 h using EPMA (a) BSE image, (b) Ni, (c) Al, (d) Cr, (e) W, and (f) O
3.3.2 Short duration exposure up to 1 hour for salt-coated samples (initiation stage)
The cross-sectional microstructure of 3MC sample corresponding to 30 minutes. exposure is shown in Figure 6(a). The deposited salt layer can be seen on the surface of the sample. Though appreciable degradation of the superalloy is not detected during 30 minutes, the dispersion of fine dark particles is observed in the sub-surface regions up to a depth of about 5 µm (Figure 6(a)). The magnified view of such a region is shown in the inset of Figure 6(a). The EDS spectrum suggests that the dark particles correspond to the sulfide phase (Figure 6(b)). The sulfide phases at the corrosion front are rich in Cr, as it would be shown later in Figure 11. After exposure for 1 hour, the deposited salt layer has completely reacted and the superalloy exhibits significant surface degradation (Figure 6(c)). The degraded zone near the surface of the sample is constituted of several layers and the overall thickness is about 35 µm. Cracking and spallation of the outer layers are evident. Oxidation along dendritic boundaries can be seen in Figure 6(c). These boundaries are evident from the presence of carbide phases (seen as a bright phase in Figure 6(c)), which is typical for cast superalloys.[32,33] The interface between the surface degraded zone and the unaffected superalloy substrate is marked by the presence of dispersion of sulfide phases (Figure 6(c)).

(a) BSE micrograph for 3MC specimen after 30 min of exposure, (b) typical EDS spectrum of fine dark phase dispersed in sub-surface region and marked as sulfide particles, (c) micrograph after 1 h of exposure. Inset in (a) shows the magnified image for 30 min of exposure
From the above observations, it is ascertained that the hot corrosion initiates by the formation of a fine dispersion of sulfide phases in the sub-surface regions, which occurs much before the formation of the corrosion scale (Figure 6(a)). With the increase in thermal exposure up to 1 hour, a thick corrosion scale forms and the extent of sulfide phase dispersion (Figure 6(c)) increases (Figure 6(a)). The inward propagation of the corrosion front into the substrate alloy is evident by comparing Figures 6(a) and (c). Therefore, it can be considered that the transition from initiation stage to propagation stage of hot corrosion occurs between 30 minutes and 1 hour for DS CM247LC superalloy under the test conditions used in the present study.
3.3.3 Long duration exposure of salt-coated samples (propagation stage)
As typically shown in Figure 7(a) for 3MC sample, 20 hours of exposure leads to the formation of about 340 µm thick corrosion scale on the superalloy sample. Based on the phase contrast, the corrosion scale can be categorized into three zones, namely Z1, Z2 and Z3, as marked in Figure 7(a). The three zones are indicated in Figure 7(a). The outer zone (Z1) is porous and constituted primarily of oxides (Figure 7(b)). Based on EPMA elemental maps (Figures 8(b) through (f), (h), (i)) and EDS analysis (Figure 7(b)), it is evident that the outer zone (Z1) is enriched in oxides of Ni, Cr, Co, Al, W and Ta. Oxides of minor elements i.e., Hf, Ti and Mo are also dispersed in the Z1 layer (Figures 8(j) through (l)). The dispersion of a bright phase in Z1 corresponds primarily to W, Ta, Hf oxides (Figures 7(a) and 8(h) and (i)). The presence of Na is also detected in Z1 (Figure 8(m)). Although the Z1 layer is lean in S, enrichment of S is observed near the interface between Z1/Z2 layers (Figure 8(g)).

3MC specimen after 20 h of exposure. (a) BSE micrograph of the hot corroded region, (b) EDS spectrum for Z1 region


Elemental maps for 3MC sample after exposure for 20 h. (a) BSE image, (b) Ni, (c) Co, (d) Al, (e) Cr, (f) O, (g) S, (h) W, (i) Ta, (j) Hf, (k) Ti, (l) Mo, and (m) Na
The intermediate zone of the corrosion scale (Z2) consists of islands of metal surrounded by oxides (dark phase), as evident from the magnified image in Figure 9(a) and the EPMA maps (Figure 8). The EDS analysis (Figure 9(b)) and the elemental maps (Figure 8) of the metal islands confirm the absence of O, Cr, Al and presence of Ni, Co, W, Ta and Hf in these islands. Cr and Al from these metal islands have been consumed for the formation of Cr2O3 and Al2O3 surrounding the islands. This can be confirmed from the corresponding EDS spectra (Figure 9(c)) and the EPMA elemental maps (Figure 8). Oxides of Ni, Co, W, Ta, Hf, and Mo are also detected along with the abovementioned Al2O3 and Cr2O3 (Figure 9(c)). A significant amount of coarse W-oxide phase along with Ta, Hf and Ti oxides is observed at the Z1/Z2 interface (Figures 10(i) through (k)). The Z2/Z3 interface is enriched with Hf, Ti and Mo (Figures 10(j) through (l)).

3MC specimen after 20 h of exposure. (a) BSE micrograph for Z2 region, (b) EDS spectrum for metal islands, and (c) EDS spectrum for dark oxide around metal islands

3MC specimen after 20 h of exposure. (a) BSE micrograph for Z3 region, (b) EDS spectrum for sulfide particles, (c) BSE micrograph showing sulfide-undercutting of substrate, (d) EDS spectrum for metal islands in Z3 region, and (e) typical EDS spectrum for unaffected substrate below sulfide particles
The innermost zone of the corrosion scale (indicated as Z3 in Figure 7(a)) exhibits the presence of oxide and sulfide phases (Figure 10). The oxide phase is rich in Al along with a minor amount of Cr (Figures 8 and 11) and it is present at the outer regions of Z3. Figure 10(b) shows that the sulfide phase is rich in Cr and it is detected at the inner region of Z3 i.e., close to Z3/substrate interface. The penetration of the sulfide phase into the superalloy and the presence of sulfide phase at the Z3/superalloy interface is well apparent from Figures 10(a) through (c) and the EPMA elemental maps (Figure 8(g)). Mo is also present near the sulfides (Figures 8(m), 10(b)). The metal islands in Z3, similar to those in Z2, are devoid of O and contain significantly less Cr and Al (Figure 10(d)) than that of the unaffected superalloy substrate (Figure 10(e)). The loss of Al and Cr from the metal islands can be ascribed to the formation of sulfide and oxide phases at the boundaries. The sulfide region at the corrosion scale/superalloy interface is an indicator of the depth of corrosion damage in the superalloy. The superalloy substrate below the corrosion front exhibits the same composition as that of the unaffected superalloy and no O and S are detected in these regions (Figure 10(e)).

(a) BSC micrograph for 3MC exposed for 20 h, EPMA elemental map for (b) O, (c) Al, (d) Cr, and (e) S. The arrows containing solid lines, short-dotted lines, and long-dashed lines indicate sulfides, oxides, and the oxide converted from sulfide, respectively
Careful examination of the EPMA elemental maps corresponding to the Z3 region suggests sulfide-oxide conversion plays an important role in the degradation of the superalloy during hot corrosion. The conversion is shown in Figure 11 and indicated by three different arrow types. The gray regions in the BSE image in Figure 11(a), which are typically indicated by the solid line arrows, contain Cr-sulfides as evidenced by the enrichment in Cr and S (Figures 11(d) and (e)). The channels with dark appearance in the BSE image and indicated by short-dotted arrows (Figure 11(a)), are predominantly rich in the oxide of Al and also contain Cr-oxide to some extent (Figures 11(b) through (e)). Formation of thin oxide at the interface of sulfide region and oxide channel, indicated by the long-dashed arrows (Figures 11(a) through (c)), suggests the conversion of the sulfide to the oxide phase and the subsequent growth of the oxide channels in place of sulfides. The presence of sulfides with no oxides near the Z3/superalloy interface and presence of oxides with no sulfides away from the Z3/superalloy interface (towards the outer layer Z2), corroborates the sulfide-oxide conversion mechanism for hot corrosion of the superalloy.
The cross-sectional microstructure of 3MC specimens corresponding to longer exposure durations reveals similar degradation features described above for the 3MC specimen exposed for 20 hours. For example, the three zones Z1, Z2, and Z3 can be clearly seen in the microstructure corresponding to 80 hours of exposure for 3MC sample, as shown in Figure 12(a). The features of the corrosion scale remain unaffected even with the increase in the amount of the initially deposited salt, which is evident from the similar multilayered cross-sectional microstructures for the 3MC (Figure 12(a)) and 12 MC samples (Figure 12(b)) after thermal exposure for 80 hours.

BSE micrograph for 3MC specimens after 80 h of exposure: (a) 3MC and (b) 12MC. Both the specimens show three-layered corrosion scale
4 Discussion
The significantly higher weight gain of DS CM247 superalloy during hot corrosion as compared to that during oxidation (i.e., without salt deposit) indicates accelerated material degradation in the presence of molten salt. Irrespective of the amount of initial salt deposit, the samples exhibit a rapid weight gain (Figure 2), concomitant reduction in the unaffected cross-sectional area, and complete degradation during 500 to 1000 hours of exposure in the air at 950 °C (Figure 4(b)). The change in specific weight is insignificant after 500 hours of exposure (Figure 2) for all the three salt conditions. The plateau in the specific weight change vs time plot after 500 hours can be ascribed to the significantly less remnant cross-section of the samples available for further reaction during exposure for durations exceeding 500 hours. As evident from Figure 4(b), the unaffected cross-sectional area is less than 15 and 5 pct of the original cross-section after 500 hours of exposure for 3MC and 12 MC samples, respectively. Therefore, degradation of most of the metal occurred during 500 hours of exposure and there was less metal available for further degradation which manifests in small and relatively insignificant weight change after 500 hours. It is demonstrated that thin salt film is detrimental enough to cause extensive damage to the superalloy. The present study shows that catastrophic degradation of the alloy, caused by pure Na2SO4 salt film, does not cease unlike that reported earlier by Gurrappa[26] and Tsao et al.[28] The degradation is self-sustained and leads to the complete conversion of the alloy into corrosion products. The complete corrosion of the entire section of the specimen and its disintegration after 1000 hours of exposure is clearly evident from Figure 1(d).
Based on the results presented in Section III, the mechanism of hot corrosion-induced degradation of DS CM247 superalloy under molten Na2SO4 is described in the following paragraphs and illustrated schematically in Figure 13. Complete conversion of the metallic alloy into corrosion product in the presence of a thin un-replenished molten salt film in air, akin to that adopted in the present study, is feasible only if the alloy degrades by a self-sustaining mechanism. The self-sustainable behavior of hot corrosion degradation can be explained using the well-known ‘Rapp–Goto criterion’.[34] This criterion proposes that accelerated attack due to molten salt is sustained through a dissolution-precipitation process if the solubility of the protective oxide scale formed on substrate surface has a negative gradient at the oxide/salt interface. The negative gradient of solubility implies that solubility of oxide is higher at the oxide/salt interface than at the salt/air interface. Such negative solubility gradient causes dissolution of the oxides at the oxide/salt interface and their re-precipitation near the salt/air interface as a porous and non-protective oxide scale. The Rapp–Goto criterion is expressed as[34]:

Schematic showing the proposed mechanism for hot corrosion of CM247LC alloy in the presence of molten Na2SO4 salt film. Different steps of the mechanism are labeled in the schematic from (a) to (j). The initiation stage is represented by steps (a) through (f) and the propagation stage is represented by steps (g) through (j). Magnified view of the metal island in the Z2 layer shown in the inset of step (j) indicates role of sulfidation-oxidation in the conversion of metal island into porous oxide scale
where ‘S’ is the solubility of oxide in the molten salt and ‘x’ is the distance from the oxide/salt interface with x = 0 representing the oxide/salt interface.
At the test temperature of 950 °C, the Na2SO4 salt film forms a molten salt film on superalloy surface (Figure 13(a)) by virtue of its melting temperature of 884 °C, as ascertained from DSC measurements. The equilibrium in the Na2SO4 melt is shown in Eq. [2] as an acid-base reaction, with Na2O (or O−2) as base and SO3 as acid.[14] The acid-base chemistry in the molten salt can be expressed as indicated in Eq. [3] at 927 °C.[13] The basicity of the melt is defined by − log \( \left( {\text{a}_{{{\text{Na}}_{2} {\text{O}}}} } \right) \) similar to that of pOH in aqueous solutions.[13,14]
For understanding the changes in the molten salt deposit due to its interaction with the substrate, an equivalent reaction can be written as given in Eq. [4][19]:
Oxygen (O) in the as-deposited molten salt reacts with the underlying superalloy to form protective oxide scale (Figure 13(b)) of Al2O3, Cr2O3 and NiAl2O4 (NiO.Al2O3).[31] The O availability from the atmosphere for the formation of oxide scale at salt/superalloy interface is negligible since the O transport across the molten Na2SO4 salt film has reported to be low.[19,35] The formation of oxides at salt/alloy interface during the initial stages of hot corrosion in Ni-Al, Ni-Cr and Ni-Cr-Al alloys under molten Na2SO4 salt film has also been reported by Goebel and Pettit.[20] Therefore, the low weight change of 0.5 mg cm−2 during the initiation stage of hot corrosion for 30 minutes (Figure 3) can be ascribed to the formation of a protective oxide scale at the salt/superalloy interface. The as-deposited salt is neutral at the initial stages and does not affect the oxidation behavior of the alloy, which is evident from the similar weight change of the samples with and without the salt deposit during the initial 30 minutes of thermal exposure (Figure 3).
The formation of oxides on the superalloy surface causes a decrease in the O concentration which leads to an increase in the sulfur (S) concentration in the molten salt according to Eq. [4]. The high concentration of S in the molten salt induces diffusion of S into the superalloy through the oxide film and its subsequent reaction with Cr to form Cr-sulfide precipitates in the alloy beneath the oxide film (Figure 13(c)). The stable sulfide phases in the Cr-S system can be denoted as CrSx where x is in the range of 1 to 1.5.[36,37] Sulfide formed in this manner is seen as the fine dispersed phase at the sub-surface regions of the superalloy during the initial stages of corrosion (Figure 6). The diffusion of S from molten salt and formation of sulfides at the corrosion front is also reported during hot corrosion in other Ni-base superalloys such as Nimonic 100,[8] B1900,[38] IN738,[39] Nimonic 105[40] and Udimet.[41]
The formation of oxides and sulfides leads to the depletion of O and S from the molten salt. It changes the basicity of the molten salt (Figure 13(c)) controlled by \( a_{{{\text{Na}}_{2} {\text{O}}}} \) in the melt (Eq. [4]). This leads to the rapid dissolution of the oxides formed at salt/superalloy interface in the molten salt through basic fluxing mechanism. The dissolution reactions can be written as[19]:
A change in the basicity of molten salt due to the depletion of O and S occurs in the vicinity of the salt/oxide interface, whereas the basicity of the molten salt away from salt/oxide interface (or near the salt/air interface) remains unaffected. As a result, the solubility of oxide in molten salt near the salt/oxide interface becomes much higher than the solubility of oxide in molten salt near salt/air interface. Based on the Rapp–Goto criterion,[34] such a negative gradient in the solubility of oxide leads to the dissolution of oxide into the salt near the salt/oxide interface and its re-precipitation near salt/air interface in a porous form (Figure 13(d)). The re-precipitated porous oxide scale does not provide protection to the superalloy against oxidation as well as corrosion attack. The dissolution of the oxides at salt/oxide interface results in the disintegration of the protective oxide scale (Figures 13(e) and (f)) and exposes the underlying superalloy to the molten salt causing rapid degradation, i.e., the transition from initiation stage to the propagation stage (Figure 3). During the propagation stage, rapid dissolution and re-precipitation of oxides of superalloy elements such as Al, Cr, Ni, and Co occurs.
Formation of W-rich region at Z1/Z2 interface (Figures 8(h) and 9(a)) was observed in all the specimens irrespective of the salt deposit amount and duration of exposure. It suggests that W plays an important role in the degradation of CM247LC alloy. The formation W-oxide and its dissolution into the salt can be represented as follows:
Na2WO4, which forms by the reaction of WO3 with molten Na2SO4 salt (Eq. [9]), remains in a molten state as the melting point of Na2WO4 i.e., 696 °C is below the test temperature of 950 °C.[42] Molten Na2WO4 decreases the basicity of the molten salt and renders it more acidic[42,43] (Figure 13(g)). Acidic Na2SO4-Na2WO4 melt leads to accelerated degradation of the material by alloy-induced fluxing. The self-sustaining nature of degradation in the present alloy confirms the role of alloy-induced acidic fluxing which is known to continue unabated.[20] Fryburg et al. explained the self-sustainable behavior of alloy-induced acidic fluxing by the repetition of the oxide dissolution reactions and reverse of these reactions.[38]
The reactions pertaining to acid fluxing and subsequent dissolution of NiO, Al2O3, and Cr2O3 into the molten salt are represented below:
The oxides dissolve in the molten salt through the abovementioned reactions and re-precipitate at the salt/air interface as porous and non-protective oxides. This is evident from the porous outer zone (Z1) of the corrosion scale (Figure 7(a)). Thus, the hot corrosion mechanism for CM247LC alloy transitions from basic fluxing during the initial stages (30 to 60 minutes) to alloy-induced acidic fluxing in the later stages of exposure. Such a changeover of fluxing mechanism from basic to acidic has also been reported for Mo containing B1900 superalloy by Fryburg et al.[38] and Giggins et al.[43] Ta present in the alloy is expected[44,45] to show beneficial effect on hot corrosion resistance of the alloy against molten salt. The oxide of Ta i.e., Ta2O5 binds with Na2O and forms solid NaTaO3. Preferential formation[44,45] of solid NaTaO3 prevents the formation of molten Na2WO4 and Na2MoO4, which provides some resistance against alloy-induced acidic fluxing. However, the degradation mechanism is primarily governed by detrimental effects of W because the alloy has higher W content than Ta.
The role of minor elements, i.e., Mo, Ti and Hf, in degradation of the alloy could not be ascertained precisely from the present study because these elements are present in small quantities (< 1.5 wt pct). However, the role of the above elements in hot corrosion can be expected to be similar as that reported in References 1, 19,20, 46 through 48. Mo is known to have detrimental effect on hot corrosion resistance of the alloy and exhibits same effect as W.[1,19,20] Similar to that of W, the oxide of Mo dissolves in the molten salt and increases the acidity of the molten salt, which makes the salt more aggressive. Limited studies are reported regarding the effects of Ti and Hf on hot corrosion behavior of Ni-based superalloy. Smith and Lewis[46] reported that Ti is beneficial in enhancing the corrosion resistance of the alloy in conjunction with Cr and Al, and expressed the effect of Ti in terms of ‘chromium equivalent’ (CE = pctCr + 0.7 pctAl + l.5 pctTi.). Leytan[47] reported that Hf has detrimental effect against Type-I hot corrosion, whereas Shimko[48] reported improved resistance against Type-II hot corrosion for Hf content of up to 2 wt pct.
As mentioned earlier in Section III–C–3, the sulfides found prominently in the corrosion product (Figures 6, 8, 10(a) through (c)) suggest a secondary sulfidation-oxidation mechanism operating simultaneously with the fluxing mechanism. The sulfides are observed at the corrosion front in all the samples corresponding to the three salt conditions, i.e., 3MC, 7MC and 12MC, irrespective of the duration of exposure. Concomitantly, the dark channels in Z3 away from the corrosion front contain oxides of Al, Cr and are depleted in S (Figures 8 and 11). The oxidation of sulfides (Figure 13(h)) initiates at the peripheral regions of the sulfides (Figure 11), which causes the formation of Cr-oxide, Al-oxide and concomitant depletion of Cr, Al in the metal island regions in the Z3 layer (Figure 11(d)). Oxidation of sulfides releases S which diffuses inward into the superalloy and forms fresh sulfide phases at the corrosion front. The process of sulfidation followed by oxidation continues unabated as the released S sustains the formation of fresh sulfides. The role of S in the repeated formation of sulfides and oxides is supported by the study carried out by Tedmon and Seybolt on the oxidation behavior of chromium sulfides,[49] which reports that the S released during oxidation of chromium sulfides diffuses into the alloy rather than evolving as sulfur oxide gas.
Apart from participating in the sulfidation-oxidation mechanism, the S assists the disintegration of the superalloy by sulfide-undercutting (Figures 10(a) and (c)). Undercutting of the alloy due to oxidation has been reported by Das et al. for CM247 superalloy[31] and Giggins and Pettit for Ni-Cr-Al alloys.[50] It is evident from the present study that undercutting, termed as sulfide-undercutting, takes place in hot corrosion as well and plays a crucial role in accelerating hot corrosion degradation. The inner portion of the corrosion product marked as Z3 in Figure 7(a), is actually a portion of the substrate in which numerous CrSx regions have formed (Figure 10(a)). While these regions are of various shapes and sizes, some of them are very large and elongated. The diffusion of S into the substrate causes the formation of these sulfide particles in zone Z3. It is apparent from Figure 10 that many of the sulfide particles have undercut into the substrate causing removal/near-removal of pieces of the substrate and their embedment into the oxide scale (Figure 13(i)). An illustration of sulfide-undercutting is shown in Figure 10(c), which shows the formation of isolated metal islands surrounded by the continuous network of sulfide phase.
With continued oxidation of sulfides and subsequent acidic fluxing of oxides, the metal islands in Z3 eventually become part of Z2 layer and fresh Z3 layer is formed above the superalloy (Figure 13(j)). The depletion of Cr and Al in the metal islands occurs due to their reaction with S and O to form Cr-sulfide and, Cr and Al oxides phases at the boundaries (Figure 10(d)). Subsequently, due to their depleted Al and Cr levels, the metal islands are converted into porous oxide and added to Z1 layer (Figure 13(j)). The present study also indicates that the metal islands, primarily constituted of Ni-Co and present in Z2 layer, do not straightaway get converted into porous Ni and Co oxides but via an intermediate step of sulfidation, this is corroborated by the enrichment of S in the outer regions of the Z2 layer, i.e., at the Z1/Z2 interface (Figure 8(g)). Close examination of the interface between Ni-Co metal islands and surrounding oxide scale revealed that metal islands are first converted into sulfides and then into their respective porous oxides (magnified view of metal island in Figure 13(j)). Therefore, the sulfidation-oxidation mechanism contributes to the final conversion of the metal islands into the porous oxide layer.
From the above discussion, it is established that the extensive degradation of CM247LC superalloy and its complete conversion into corrosion product is caused by two corrosion mechanisms operating in tandem: (i) fluxing and (ii) sulfidation-oxidation. These two mechanisms can occur independent of each other. Sulfidation is known to occur in S containing environment and does not necessarily require molten salt which is, however, essential for fluxing mechanisms. Similarly, the fluxing mechanisms do not require sulfidation as a pre-requisite. Bornstein and De Crescente[51] showed experimentally that S was non-essential for the fluxing mechanism to cause rapid degradation by demonstrating accelerated hot corrosion using non-sulfur containing salts such as Na2CO3 or Na2NO3. Therefore, when both mechanisms are simultaneously active, which is the case in the present study, it becomes essential to highlight the effect of one mechanism on the other. Based on the discussion, the interaction between fluxing mechanism and sulfidation-oxidation mechanism is proposed in the following manner. The sulfidation-oxidation mechanism assists the fluxing mechanism by neutralizing the Cr, which is known to show resistance against fluxing, present in the alloy by forming Cr-sulfides. The ability of Cr to resist the attack against the molten salt comes from its ability to establish a positive solubility gradient in the molten salt. Multiple valance states of Cr make the solubility of Cr2O3 in molten salt oxygen partial pressure dependent.[9] Higher oxygen partial pressure near salt/gas interface and lower oxygen partial pressure near salt/oxide interface leads to the positive solubility gradient for Cr2O3. The positive solubility gradient allows dissolution of Cr2O3 into the molten salt to its saturation level without re-precipitation. Molten salt saturated with Cr2O3 cannot dissolve additional Cr2O3, thus, stopping the molten salt attack. Depletion of Cr through the sulfidation-oxidation mechanism renders the Cr-depleted alloy prone to rapid fluxing attack in presence of the molten salt. Additionally, the porous oxide channels formed in the alloy due to sulfidation-oxidation and sulfide-undercutting mechanisms allow infiltration of molten salt deep into the substrate. In reciprocation, the fluxing mechanism generates additional S from Na2SO4 present in Na2SO4-Na2WO4 melt according to Eq. [4]. This additional S diffuses into the alloy for participating in the ongoing sulfidation-oxidation at the corrosion front. The rapid degradation and complete conversion of the CM247LC superalloy into corrosion product in presence of molten Na2SO4 salt film occurs due to the synergy of sulfidation-oxidation mechanism and fluxing mechanism.
5 Summary
The present study describes the Type-I hot corrosion behavior of DS CM247LC superalloy under molten Na2SO4 salt in the air at 950 °C. The alloy undergoes accelerated degradation under molten N2SO4 as compared to that of oxidation in air at 950 °C. The corrosive degradation of the alloy increases with the increase in the duration of exposure. Low levels of initial salt deposit, such as 3 to 4 mg cm−2, without further salt replenishment, is detrimental enough to cause complete degradation of the alloy after about 500 hours of exposure. The progress of degradation with time can be divided into two stages, namely, initiation stage and propagation stage. The initiation stage occurs during the initial 30 minutes of exposure. The Al and Cr-oxides formed by reaction with O from the molten salt impart low corrosion rate during the initial exposure of the alloy for 30 minutes. However, the propagation stage starts when the basic fluxing of the oxides by the molten salt causes significant enhancement of the corrosion rate. The degradation becomes self-sustained and continues unabated as the fluxing mechanism changes from basic fluxing to alloy-induced acidic fluxing during the propagation stage. The severe degradation of the alloy is caused by the fluxing, sulfidation-oxidation, and sulfide-undercutting mechanisms.
References
F. Pettit, Oxid. Met., 2011, vol. 76, pp. 1-21.
C. G. McCreath, Mater. Sci. Technol., 1987, vol. 3, pp. 494-500.
V. I. Hanby, J. Eng. Power., 1974, vol. 96, no. 2, pp. 129-133.
M. A. De Crescente, N. S. Bornstein, Corrosion, 1968, 24, pp. 127-133.
R. A. Rapp, Y. S. Zhang, JOM, 1994, vol. 46, no. 12, pp. 47-55.
R. A. Rapp, Pure Appl. Chem., 1990, vol. 62, no. 1, pp. 113-122.
J. A. Goebel, F. S. Pettit, Metall. Mater. Trans. B, 1970, vol. 1, pp. 1943-1954.
J. Stringer, Mater. Sci. Technol., 1987, vol. 3, no. 7, pp. 482-493.
P. Hancock, Mater. Sci. Technol., 1987, vol. 3, no. 7, pp. 536-544.
N. Eliaz, G. Shemesh, R. M. Latanision 2002 Eng. Fail. Anal. vol. 9, no. 1, pp. 31-43.
T. S. Sidhu, S. Prakash, R. D. Agrawal, Curr. Sci., 2006, vol. 90, no. 1, pp. 41-47.
B. Grégoire, X. Montero, M. C. Galetz, G. Bonnet, F. Pedraza, Corr. Sci., 2019 vol. 155, pp. 134-145.
R. A. Rapp, Corr. Sci., 2000, vol. 44, pp. 209-221.
D. J. Young (2016) High Temperature Oxidation and Corrosion of Metals. Elsevier, Amsterdam.
N. Birks, G. H. Meier, F. S. Pettit, JOM, 1987, vol. 39, no. 12, pp. 28-31.
G. Meier, Mater. Sci. Eng. A, 1989, vol. 120–121(1), pp. 1-11.
Z. Rizhang, G. Manjiou, Z. Yu, Oxid. Met., 1987, vol. 27, pp. 253-265.
N. S. Bornstein, JOM, 1996, vol. 48, no. 11, pp. 37-39.
N. Birks, G. H. Meier, F. S. Pettit, Introduction to the High-Temperature Oxidation of Metals, 2nd ed., Cambridge University Press, Cambridge, 2006, pp. 205-252.
J. A. Goebel, F. S. Pettit, G. W. Goward, Metall. Mater. Trans. B, 1973, vol. 4, pp. 261-278.
G.L. Erickson, K. Harris, R. E. Schwer: in Proc. of ASME 1985 International Gas Turbine Conference and Exhibit, Houston, Texas, 1985, paper no. 85-GT-107, pp. 1–10.
K. Harris, G.L. Erickson, R. E. Schwer: in 5th Int. Symp. Super- alloys, Seven Springs, PA. TMS Proceedings, 1984, pp. 221–30.
G.L. Erickson, K. Harris: in Proc. of the 5th Conf. on Materials for Advanced Power Engineering (Liege, Belgium), 1994, Pt. 2, pp. 1055–1074.
R. K. Rai, J. K. Sahu, P. S. M. Jena, S. K. Das, N. Paulose, C. D. Fernando, Mater. Sci. Eng. A, 2017, vol. 705, pp. 189-195.
M. Z. Alam, D. V. V. Satyanarayana, D. Chatterjee, R. Sarkar, D. K. Das, Mater. Sci. Eng. A, 2015, vol. 641, pp. 84-95.
I. Gurrappa, Oxid. Met., 1999, vol. 51, no. 5, pp. 353-382.
J. Sumner, A. Encinas-Oropesa, N. J. Simms, J. E. Oakey, Mater. Sci. Technol., 2013, vol. 29, no. 7, pp. 813-821.
T. K. Tsao, A. C. Yeh, C. M. Kuo, H. Murakami, Entropy, 2016, vol. 18, no. 2, p. 62.
BS ISO 21608:2012, Edition 1, ICS 77.060, 2012.
ISO 26146:2012(E), Edition 1, 2012.
D. K. Das, V. Singh, S. V. Joshi, Mater. Sci. Technol., 2003, vol. 19, pp. 695-708.
T. M. Pollock, S. Tin, J. Prop. Power, 2006, vol. 22, no. 2, pp. 361-374.
A. Basak, S. Das, J. Alloys Compd., 2017, vol. 705, pp. 806-816.
R.A. Rapp, K.S. Goto: in Proc. of the Second International Symposium on Molten Salts, eds. J. Braunstein and J.R. Selman, Electrochemical Society, Proceedings, vol. 81–10, 1981, pp. 159–77.
B. Grégoire, X. Montero, M. C. Galetz, G. Bonnet, F. Pedraza, Corr. Sci., 2018, vol. 141, pp. 211-220.
J. X. Chang, D. Wang, G. Zhang, L. H. Lou, J. Zhang, Corr. Sci., 2017, vol. 117, pp. 35-42.
D. J. Young, W. W. Smeltzer, J. S. Kirkaldy, J. Electrochem. Soc., 1973, vol. 120, pp. 1221-1224.
G. C. Fryburg, F. J. Kohl, C. A. Stearns, J. Electrochem. Soc., 1982, vol. 129, no. 3, pp. 571-585.
G. C. Fryburg, F. J. Kohl, C. A. Stearns, J. Electrochem. Soc., 1984, vol. 131, no. 12, pp. 2985-2997.
K. R. Peters, D. P. Whittle, J. Stringer, Corr. Sci., 1976, vol. 16, pp. 791-804.
A. K. Misra, Oxid. Met., 1986, vol. 25, pp. 129-161.
P. Kofstad, High Temperature Corrosion, Elsevier Applied Science, London, 1988, pp. 465-510.
C.S. Giggins, F.S. Pettit: Pratt & Whitney Rept. No. PWA-FR-11545, June 1979, https://apps.dtic.mil/dtic/tr/fulltext/u2/a072645.pdf. Accessed 22 Sept 2020.
G. C. Fryburg, C. A. Stearns, F. J. Kohl (1977) J. Electrochem. Soc. 124: 1147-1148.
J. X. Chang, D. Wang, T. Liu, G. Zhang, L. H. Lou, J. Zhang, Corr. Sci., 2015, vol. 98, pp. 585-591.
H. Lewis, R.A. Smith: First International Congress on Metallic Corrosion (April 1961), Butterworths, London, 1962, pp. 202–14.
K.N. Soto Leytan: Ph.D Thesis, University of California, Irvine, 2019, https://escholarship.org/uc/item/83c2c2x0. Accessed 22 Sept 2020.
M.J. Shimko: Master’s Thesis, Naval postgraduate school, Monterey, California, 1983, http://hdl.handle.net/10945/19861. Accessed 22 Sept 2020.
C. S. Tedmon Jr., A. U. Seybolt, Corr. Sci., 1968, vol. 8, pp. 125-132.
C. S. Giggins, F. S. Pettit, J. Electrochem. Soc., 1971, vol. 118, no. 11, pp. 1782-1790.
N. S. Bornstein, M. A. DeCrescente, Metall. Mater. Trans. B, 1971, vol. 2, no. 10, pp. 2875-2883.
Acknowledgments
The authors would like to convey their sincere gratitude to Director, DMRL for his permission to publish the present work. The authors acknowledge the assistance provided by EMG and SFAG groups of DMRL. This research work is funded by the Defence Research and Development Organization (DRDO).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted July 5, 2020; accepted October 11, 2020.
Rights and permissions
About this article

Cite this article
Kumawat, M.K., Parlikar, C., Alam, M.Z. et al. Type-I Hot Corrosion of Ni-Base Superalloy CM247LC in Presence of Molten Na2SO4 Film. Metall Mater Trans A 52, 378–393 (2021). https://doi.org/10.1007/s11661-020-06068-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-020-06068-6