
Abstract
Despite a strong preclinical rationale for targeting the insulin-like growth factor (IGF) axis in cancer, clinical studies of IGF-1 receptor (IGF-1R)-targeted monotherapies have been largely disappointing, and any potential success has been limited by the lack of validated predictive biomarkers for patient enrichment. A large body of preclinical evidence suggests that the key role of the IGF axis in cancer is in driving treatment resistance, via general proliferative/survival mechanisms, interactions with other mitogenic signaling networks, and class-specific mechanisms such as DNA damage repair. Consequently, combining IGF-targeted agents with standard cytotoxic agents, other targeted agents, endocrine therapies, or immunotherapies represents an attractive therapeutic approach. Anti-IGF-1R monoclonal antibodies (mAbs) do not inhibit IGF ligand 2 (IGF-2) activation of the insulin receptor isoform-A (INSR-A), which may limit their anti-proliferative activity. In addition, due to their lack of specificity, IGF-1R tyrosine kinase inhibitors are associated with hyperglycemia as a result of interference with signaling through the classical metabolic INSR-B isoform; this may preclude their use at clinically effective doses. Conversely, IGF-1/IGF-2 ligand-neutralizing mAbs inhibit proliferative/anti-apoptotic signaling via IGF-1R and INSR-A, without compromising the metabolic function of INSR-B. Therefore, combination regimens that include these agents may be more efficacious and tolerable versus IGF-1R-targeted combinations. Herein, we review the preclinical and clinical experience with IGF-targeted therapies to-date, and discuss the rationale for future combination approaches as a means to overcome treatment resistance.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 The IGF Axis and Its Role in Tumor Biology
1.1 IGF: Dramatis Moleculae
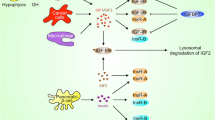
Insulin-like growth factor (IGF) signaling plays an important role in regulating growth and development in normal human tissues by promoting cellular proliferation and differentiation and preventing apoptosis [1, 2]. The IGF axis comprises insulin and two related ligands, IGF ligands 1 and 2 (IGF-1 and IGF-2) that regulate cellular processes by interacting with specific cell-surface receptors (Fig. 1) [1, 5]. The IGF-1 receptor (IGF-1R) is a heterotetrameric receptor with two extracellular, ligand-binding domains (alpha subunits) and two transmembrane beta subunits that contain the kinase domain; the alpha and beta domains are linked by disulfide bonds [6]. Along with the insulin receptor (INSR), IGF-1R is a member of the receptor tyrosine kinase (RTK) class 2 family of receptors (insulin receptor family) [1, 3]. IGF-1R binds the IGF ligands with varying affinities depending on the cell type and experimental conditions, both IGFs binding with higher affinity than insulin. IGF-2 also binds to INSR-A, a fetal isoform that is overexpressed in some tumors, and to IGF-2 receptor (IGF-2R), a structurally unrelated receptor that lacks tyrosine kinase activity and serves as a scavenger for circulating IGF-2 [7].

Components of the insulin/IGF axis. The IGF axis consists of ligands (insulin, IGF-1, and IGF-2), receptors (INSR, IGF-1R, IGF-2R, and IGF-1R/INSR hybrid receptors), IGFBPs 1 to 7, and IGFBP proteases. The IGF ligands bind their receptors and binding proteins with high affinity. IGFBPs bind tightly to IGF ligands, influencing binding to their receptors; IGFBP proteases cleave the IGFBPs into fragments with lower affinity for the IGF ligands, thereby increasing free IGF-1 and IGF-2 bioavailability. IGF-1/IGF-2 and insulin can cross-bind to each other’s receptor, albeit with much weaker affinity than that for the preferred ligand. Activation of the INSR-B isoform regulates glucose metabolism, while activation of IGF-1R, INSR-A, and IGF-1R/INSR hybrid receptors promotes cellular growth, proliferation, survival, and metastasis. IGF-2R is an unrelated monomeric receptor that acts as a scavenger for circulating IGF-2 [1, 3, 4]. IGF-1/−2 insulin-like growth factor ligand 1/2, IGFBP IGF binding protein, IGF-1R/IGF-2R type 1/type 2 IGF receptor, INSR insulin receptor
In addition to INSR-A, which binds insulin and IGF-2 with equal affinity and transmits proliferative/survival signals, INSR exists in the classical metabolic isoform, INSR-B, which binds insulin [1] and regulates glucose uptake [5, 7]. Among these components, the IGF axis includes hybrid receptors composed of IGF-1R and INSR isoforms; these IGF-1R/INSR heterodimers contain predominantly INSR-A in malignant cells, and bind IGF-1, IGF-2, and insulin [7].
The IGF system also encompasses various IGF-binding proteins (IGFBPs), IGFBP-specific proteases, and IGFBP-related peptides, which bind to and modify the activity of IGF ligands (Fig. 1) [1, 5]. Aside from their endocrine role in IGF transport, IGFBPs have many additional, IGF-independent biological functions that modulate cellular growth and survival [8]. For example, in breast cancer, IGFBP3 has been shown to interact with the epidermal growth factor receptor (EGFR) and can influence the response to DNA damage [9]. A comprehensive assessment of IGFBP biology is beyond the scope of this review. However, for more details, please refer to a 2014 review paper by Baxter [8].
1.2 The IGF Axis in Cancer
Increased expression of IGF-1R and/or circulating levels of IGF ligands has been observed in various cancers, including Ewing sarcoma, breast cancer, prostate cancer, pancreatic cancer, melanoma, and many other tumor types; this overexpression is associated with faster disease progression and a poor prognosis in some tumors [10,11,12]. Moreover, the presence of a functional IGF-1R has been shown to be essential for malignant transformation [13]. IGF-1R overexpression can result from the loss of tumor suppressors, including p53, breast cancer gene-1 (BRCA1), von Hippel-Lindau protein and Wilms’s tumor suppressor WT1 [14,15,16]. However, IGF signaling in tumor cells is driven by the presence of the ligands rather than receptor aberrations [14], and there is evidence that a low circulating IGF-1 concentration can protect against tumorigenesis [17, 18]. IGF ligands can be delivered to the IGF-1R from distant (endocrine) or nearby (paracrine) sources, with circulating free-ligand levels regulated by the different IGFBPs [2]. In addition, autocrine production of IGF ligands in some tumor cells which themselves express IGF-1R can also contribute to tumor proliferation [19, 20].
In addition to IGF-1 and IGF-1R, overexpression of IGF-2 and INSR-A is reported in a wide range of human cancers, including breast, colorectal, prostate, and lung cancers, and is also associated with a poor prognosis [21,22,23]. In preclinical studies, direct upregulation of IGF-2 in response to chemotherapy was shown to enhance the aggressive properties of castration-resistant prostate cancer (CRPC) models by activating INSR-A and/or IGF-1R/INSR-A hybrid receptors; thus, both IGF ligands can function as part of an adaptive mechanism that promotes resistance and tumorigenicity in certain cancers, during or after treatment [23, 24]. Furthermore, in tumor cells where IGF-1R and INSR are co-expressed, upregulated INSR can compensate for IGF-1R inhibition, thereby limiting sustained inhibition of downstream signaling and contributing to resistance to IGF-1R-targeted therapy [25, 26].
Recently, Josep Llovet and co-workers [27] showed that, in hepatocellular carcinoma (HCC), IGF-2 expression can be upregulated through epigenetic mechanisms, whereby increased IGF-2 mRNA and protein levels in human HCC tissues were associated with reduced methylation at the fetal IGF-2 promoters [27]. Furthermore, in mice with HCC xenografts, treatment with xentuzumab (an IGF-neutralizing monoclonal Ab [mAb]) either as a single agent or in combination with sorafenib (which is routinely used in the treatment of liver cancer) reduced tumor growth rate and improved survival compared with vehicle-treated or sorafenib-treated controls. Thus, IGF-2 appears to be an actionable oncogene target for drugs that inhibit both IGF ligands [27].
The IGF axis potentially contributes to and sustains the acquisition of several hallmarks of cancer [28]. For example, dysregulated IGF signaling is thought to contribute to an imbalance between cancer stem-cell self-renewal and differentiation, resulting in the development of non-differentiated cells with stem cell-like properties, and subsequent malignant transformation [29]. Activated IGF-1R can stabilize integrins and promote epithelial mesenchymal transition (EMT), which, in cancer, can contribute to subsequent progression to metastatic disease [30]. In addition, the IGF pathway has been implicated in DNA damage repair processes [9, 31,32,33]. Exposure to R-1507, a mAb directed against IGF-1R, downregulated the nucleotide excision repair pathway in human small cell lung cancer (SCLC) cell lines and induced chemo- and radio-sensitization to cisplatin/ionizing radiation in selected SCLC models [32]. Similarly, IGF-1R depletion or inhibition has been shown to attenuate repair of radiation-induced double strand breaks (DSBs) in prostate cancer cell lines [31, 33].
On activation of IGF-1R or IGF-1R/INSR hybrid receptors, downstream signal transduction occurs via two major pathways (Fig. 2) [5, 34]. Activation of the RAS/mitogen-activated protein kinase (MAPK) cascade results in the transcription of genes that drive cellular proliferation, such as the D-type cyclins. Activation of cyclin D/cyclin-dependent kinase 4 (CDK4) complexes leads to increased E2F-dependent transcription, via liberation of the E2F transcription factor from sequestration by the retinoblastoma protein, so driving passage through the G1/S-phase transition during cell-cycle progression [35,36,37]. In parallel, signaling via the phosphatidylinositol 3-kinase/protein kinase-B (PI3K/Akt) pathway confers protection from apoptosis [5]. In addition, Akt acts on cell-cycle regulatory proteins, inducing inhibitory phosphorylation of glycogen synthase kinase-3 beta, p21, and p27, to stabilize type-D cyclins and promote cell-cycle progression [38,39,40]. Akt also promotes cyclin D function via induction of the mammalian target of rapamycin complex 1 (mTORC1), by preventing its inhibition by tuberous sclerosis complex (TSC) proteins. This ultimately leads to ribosomal biogenesis and more efficient translation of cell-cycle progression-specific mRNAs, which are essential for cellular growth and subsequent cellular proliferation [41, 42]. Hence, both the MAPK and PI3K/Akt pathways contribute to cell-cycle progression and proliferation mediated by IGF-1R or IGF-1R/INSR hybrid receptors.

IGF-1R downstream signaling. Ligand binding to IGF-1R or IGF-1R/INSR hybrid receptors leads to phosphorylation of tyrosines that create binding sites for docking proteins including IRS 1–6 and Shc. Recruitment of IRS and Shc activates signaling via the PI3K/Akt and Ras/Raf/MAPK pathways, which regulate cellular proliferation, survival, migration, and metabolism. In addition to these pathways, interactions between IGF-1R and integrins, via scaffolding with RACK1 and FAK proteins, regulate cellular adhesion and motility [5]. Blue arrows indicate activation. Red arrows indicate inhibition. P indicates major sites of phosphorylation. Akt protein kinase B, ERK extracellular-signal-regulated kinase, IGF-1R type 1 insulin-like growth factor receptor, INSR insulin receptor, IRS INSR substrate, MAPK mitogen-activated protein kinase, MEK mitogen-activated protein kinase/Erk kinase, mTORC1 mammalian target of rapamycin complex 1, PI3K phosphatidylinositol-3-kinase, PTEN phosphatase and tensin homolog, Shc Src homology and collagen domain protein
In addition to this canonical signaling from the plasma membrane, IGF-1R can be modified by the addition of small ubiquitin-like modifier proteins (sumoylation) that enable it to translocate to the nucleus, where it can interact with chromatin and regulate gene transcription [43,44,45,46]. Nuclear IGF-1R has been shown to bind to IGF-1R promoter DNA and stimulate IGF-1R gene expression in estrogen receptor (ER)-positive breast cancer cells [47]. Finally, as well as the two main signal transduction pathways and post-translational modifications such as sumoylation, IGF-1R signaling is affected by alternative downstream mechanisms, through interactions with other plasma-membrane molecules, such as integrins and G-protein-coupled receptors [48].
Overall, it is clear that IGF signaling has the capacity to influence many of the underlying phenotypes of cancer. However, the IGF axis is only one component within a complex system of signaling networks, all of which play a part in promoting the invasive properties of cancer [49]. A large body of preclinical evidence suggests that the main function of IGF signaling in cancer is in conferring resistance to other types of anti-cancer therapy [2, 49, 50]. This is supported by the very rare occurrence of mutations in IGF-axis genes in common cancers, implying that the IGF axis is seldom the primary driver of tumorigenesis [51, 52].
In this review, we will summarize the clinical experience with different therapeutic strategies that target the IGF axis, including discussion of why dual IGF-1/IGF-2-targeting approaches and how the use of predictive biomarkers for patient enrichment might potentially improve clinical benefit. Finally, we will discuss the rationale for future combination approaches as a means to overcome treatment resistance.
2 Strategies to Target the IGF System in Cancer
2.1 IGF-1R mAbs
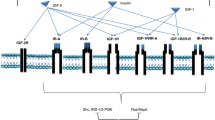
A number of therapeutic strategies that target the IGF system have been tested in a wide range of cancer types. These approaches fall into three major categories (Fig. 3). The initial strategy was to use anti-IGF-1R mAbs in order to block ligand–receptor interactions, and induce receptor internalization and subsequent degradation [21]. Many therapeutic mAbs directed against IGF-1R have been developed, and encouraging preclinical activity has been observed with ganitumab [53], figitumumab [54], dalotuzumab [67], R-1507 [55], robatumumab [56], and istiratumab, a tetravalent bispecific mAb that inhibits both IGF-1R and ErbB3 [57], in a wide range of tumor models [1]. Although IGF-1R specific, some of these agents can also induce downregulation of INSR-A or -B in IGF-1R/INSR hybrid receptors, potentially through receptor endocytosis [68]. However, these agents do not inhibit IGF-2 activation of INSR-A homodimers [65, 69].

Examples of IGF-targeted agents. Anti-IGF-1R mAbs block ligand–receptor interactions and induce receptor internalization and degradation [53,54,55,56,57,58,59]. Small molecule TKIs bind to the receptor tyrosine kinase domain and block signaling downstream of IGF-1R and INSR [60,61,62,63,64]. IGF ligand-neutralizing mAbs bind to and neutralize both IGF ligands, thereby blocking the activation of IGF-1R and INSR-A [65, 66]. Agents shown in gray have only been evaluated preclinically. Agents in italics are in ongoing clinical trials as monotherapy or in combination with other anti-cancer treatments. Clinical development of all other agents shown has been terminated. IGF-1/−2 insulin-like growth factor ligand 1/2, IGF-1R type 1 IGF receptor, INSR insulin receptor, mAb monoclonal antibody, TKI tyrosine kinase inhibitor
2.2 IGF-1R Tyrosine Kinase Inhibitors (TKIs)
An alternative approach to targeting IGF-1R signaling has been to use TKIs that inhibit RTKs. Therapeutic TKIs were developed initially as IGF-1R-specific inhibitors; however, a high level of homology in the kinase domains of IGF-1R, INSR-A, and INSR-B means that, in intact cells and in vivo, IGF-1R TKIs can affect all three [6, 70]; thus, linsitinib, BMS-754807 and KW-2450 are often described as dual IGF-1R/INSR inhibitors [60,61,62]. Each of these agents has shown promising activity in preclinical models; for example, linsitinib demonstrated superior anti-tumor activity compared with the selective anti-IGF-1R mAb MAB391 in mice bearing human tumor xenografts [25]. However, cross-reactivity with INSR-B can modulate glucose metabolism in vivo, leading to insulin resistance and hyperglycemia [21, 71, 72]. In addition, there are other known off-target effects with IGF-1R TKIs, e.g. inhibition of TrkA, TrkB, and aurora kinases by BMS-754807 [60], which may influence their therapeutic use.
2.3 IGF-1/−2 Blocking mAbs
A third therapeutic approach is to target the IGF-1 and IGF-2 ligands directly, thereby inhibiting proliferative/anti-apoptotic signaling via IGF-1R and INSR-A and their hybrid receptors, without compromising the function of INSR-B or affecting insulin action [65, 66, 73]. Thus, these agents demonstrate a lower potential for hyperglycemia compared with IGF-1R TKIs [65, 66]. In contrast with IGF-1R-specific mAbs, which can show reduced anti-proliferative activity in heterogeneous cell populations that contain a high percentage of INSR-A-expressing cells, IGF-1/IGF-2-neutralizing mAbs should also have greater anti-tumor activity in tumors that express both IGF-1R and INSR-A, or INSR-A alone [65].
Two IGF-1/IGF-2 co-neutralizing mAbs, dusigitumab (MEDI-573) [65] and xentuzumab (BI 836845) [66], have been developed. Dusigitumab shows stronger binding affinity for IGF-2 than IGF-1, while xentuzumab binds with higher affinity to IGF-1 [65, 66]. Both dusigitumab and xentuzumab have demonstrated encouraging anti-proliferative activity in vitro and in vivo in preclinical models [65, 66, 74]. Xentuzumab also binds rat and mouse IGF-1 and IGF-2, which has allowed a comprehensive characterization of its in vivo pharmacodynamic properties in both species. In rats, there was no significant toxicity following long-term exposure to xentuzumab at levels sufficient to attenuate serum IGF bioactivity and somatic growth [66].
Dusigitumab and xentuzumab are in ongoing clinical development in early-phase trials, either as monotherapy or in combination with other treatments, in metastatic hormone receptor-positive breast cancer, CRPC, and non-small cell lung cancer (NSCLC). Phase I clinical data for dusigitumab in US patients with advanced solid tumors have shown preliminary anti-tumor activity, with 13 of 39 patients in the dose-escalation cohort achieving stable disease. Dusigitumab demonstrated a favorable toxicity profile, the most common adverse events (AEs; of any cause) being decreased appetite and fatigue [75]. Similarly, dusigitumab was well tolerated in a phase I dose-escalation study in Japanese patients with advanced solid tumors, in which the best response was stable disease in four out of 10 patients [76]. Xentuzumab has also demonstrated good tolerability in three phase I dose-escalation studies using weekly or 3-weekly dosing, in patients with advanced solid tumors in the UK, Asia, and Japan. Similar to dusigitumab, the most common AEs in these trials included decreased appetite, gastrointestinal disorders and fatigue [77,78,79]. In addition, encouraging anti-tumor activity has been observed with xentuzumab, whereby two out of 48 (4%) patients in the Asian study had a partial response and a further 15 (31%) patients had stable disease [78]. In the Japanese study, there was a confirmed partial response in one of nine patients (patient with desmoid tumor) treated at the recommended phase II dose (RP2D; 1000 mg weekly), and two patients had confirmed stable disease [77].
3 Clinical Experience with IGF-Targeted Agents As Monotherapy
3.1 First Conundrum: The Absence of Meaningful Monotherapy Activity with IGF-1R-Targeted Agents
Despite a strong preclinical rationale, targeting the IGF axis has provided distinct challenges, and, with the exception of IGF-ligand-blocking mAbs, clinical development programs for most IGF-targeted agents have been compromised by failures in the clinical trial setting [80]. To date, clinical trials of single-agent anti-IGF-1R mAbs and IGF-1R TKIs have been disappointing, reporting low objective response rates and with only minor responses and disease stabilization in most tumor types (Table 1) [58, 59, 62, 81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96]. Exceptions include Ewing sarcoma, with evidence of activity in approximately 10% of patients, and soft-tissue sarcomas [81,82,83,84,85]. Recently published phase II data showed that a subset of patients with Ewing sarcoma achieved durable (>4 years) responses to the IGF-1R mAb robatumumab [97]. So far, there has been insufficient evidence of single-agent activity in common cancers, and as a result, clinical development has been terminated for most IGF-1R-targeted mAbs and TKIs.
Potential reasons for these negative clinical data with anti-IGF-1R mAbs may include an inability to sustain inhibition of proliferative/survival signaling in vivo, since these agents fail to suppress IGF-2-induced INSR-A signaling in tumor cells [73]. For IGF-1R TKIs, the risk of hyperglycemia due to co-inhibition of INSR-B may limit their ability to achieve clinically relevant IGF-1R blockade in tumor tissue [1]. Notably, neither of these limitations would be expected to apply to dusigitumab or xentuzumab based on their IGF-ligand-blocking mechanism of action [65, 66].
In addition to these limitations, resistance to IGF-targeted agents can arise from redundancy and crosstalk with other RTK signaling pathways, including EGFR, human epidermal growth factor receptor 2 (HER2), vascular endothelial growth factor receptor (VEGFR), mesenchymal epithelial transition factor (cMET), platelet-derived growth factor receptor (PDGFR), and anaplastic lymphoma kinase (ALK), as well as with steroid hormone receptors such as the androgen receptor (AR), ER, and progesterone receptor (PR) [49, 50, 63, 98,99,100,101,102,103,104,105,106,107] (Fig. 4). Furthermore, preclinical data in breast cancer suggest that inhibition of IGF-1R upregulates INSR-A, leading to IGF-2-induced amplification of Wnt and Notch signaling, which results in the expansion of stem/progenitor populations; this may lead to the development of tumors with high self-renewal potential and confer resistance to other types of anti-cancer treatment [108, 109].

Crosstalk between IGF-1R and other RTKs/steroid hormone receptors. Crosstalk between IGF-1R and other RTKs (including ALK, VEGFR, PDGFR, cMET, FGFR, EGFR, and HER2) and steroid hormone receptors (ER, AR, and PR), and compensatory activation of the MAPK and PI3K/Akt signaling pathways downstream of IGF-1R signaling can confer resistance to targeted therapies [2, 49]. Akt protein kinase B, AR androgen receptor, ALK anaplastic lymphoma kinase, cMET mesenchymal epithelial transition factor, EGFR epithelial growth factor receptor, ER estrogen receptor, ERK extracellular-signal-regulated kinase, FGFR fibroblast growth factor receptor, HER2 human epidermal growth factor receptor 2, IGF-1R type 1 insulin-like growth factor receptor, INSR insulin receptor, MEK mitogen-activated protein kinase/Erk kinase, mTOR mammalian target of rapamycin, PDGFR platelet-derived growth factor receptor, PI3K phosphatidylinositol-3-kinase, PR progesterone receptor, Shc Src homology and collagen domain protein, VEGFR vascular endothelial growth factor
3.2 Second Conundrum: The Absence of a Bona Fide Predictive Biomarker
A notable challenge has been the lack of a bona fide predictive biomarker to allow selection of patients who are most likely to benefit from IGF-targeted therapy. Although there is near-ubiquitous expression of IGF-1R in human cancers, mutations in IGF-1R are rarely reported [51, 52]. IGF-1R is, therefore, considered an “epi-driver” gene, that is, one that is expressed aberrantly in tumors, but is not the main driver of tumor development [110]. For this reason, other predictive biomarkers may be needed for patient selection in clinical trials of IGF-targeted therapies [21].
Although the IGF axis appears to be relatively unperturbed by somatic mutations, genetic studies suggest that the risk of cancer may be influenced by IGF and/or insulin gene variants. For example, non-synonymous polymorphisms in the IGF-2R gene appear to contribute to the risks of esophageal and gastric cancers [111]. Moreover, single-nucleotide polymorphisms (SNPs) in IGF-axis genes were associated with improved progression-free survival (PFS) and overall survival (OS) outcomes with chemotherapy in patients with gastric cancer [112]. A polymorphism in the 3′ untranslated region of IGF-1 was also found to be significantly associated with improved survival in patients with NSCLC [113]. In a pooled analysis that included more than 3000 cancer specimens, cancer-associated somatic copy number variations (CNVs) were detected in the IGF-1R gene region [114]. In addition, high polysomy in the IGF-1R gene was found to predict improved survival in operable NSCLC [115], suggesting that, similar to polymorphisms, CNVs could have a prognostic value in cancer. Further investigation is needed to determine whether molecular markers such as these could also serve as potential predictive biomarkers for response to IGF-targeted therapies.
A number of clinical studies have investigated possible associations of circulating free or total IGF-1 with clinical benefit of IGF-targeted agents, using serum and/or plasma-based assays, albeit with mixed results (Table 2) [116,117,118,119,120,121,122,123,124,125]. Similar to molecular markers, further research to confirm circulating free or total IGF-1 as a predictive biomarker, along with robust validation of any assay procedure(s) used, is a prerequisite for use in randomized clinical trials.
Other candidate biomarkers, either within the IGF axis or in other pathways that influence response to IGF-targeted therapies, have been investigated using various IGF-1R-targeted agents [21]. For example, preclinical and clinical evidence suggests that high expression of IGF-2 and insulin receptor substrates, and differential expression of different IGFBPs may be associated with increased sensitivity to anti-IGF-1R mAbs [122, 126, 127]. In colorectal cancer (CRC) cell lines, dusigitumab neutralized IGF-2 and induced significant tumor growth inhibition in mouse models that overexpressed IGF-2, a finding that supports evaluation of dusigitumab in CRC patients whose tumors overexpress IGF-2 [128]. Pavlicek et al. investigated correlations between genomic characteristics of cancer cell lines and figitumumab anti-proliferative activity, and found that components of the IGF pathway, including insulin receptor substrate 2 (IRS2) and IGFBP5, played a pivotal role in determining the sensitivity of tumors to single-agent figitumumab. In addition, alteration or expression of the MYB oncogene was reported to be associated with sensitivity to IGF-1R inhibitors [126]. In preclinical models of SCLC, low levels of phosphorylated extracellular-signal-regulated kinase (ERK) prior to treatment appeared to be predictive of resistance to IGF-1R inhibition [129]. With regard to clinical studies, exclusive nuclear IGF-1R was associated with response to various IGF-1R Abs in patients with sarcoma, shown by improved PFS and OS, suggesting IGF-1R nuclear translocation could also be a biomarker of IGF pathway activation [130]. However, despite these findings, there is currently a lack of any validated predictive biomarkers for patient enrichment in clinical trials of IGF-targeted agents [21].
4 Rationale for Combination Studies Using IGF-Targeted Agents
4.1 Mechanisms of Resistance
The extensive interactions between the IGF axis and other signaling networks/cellular processes inevitably lead to signaling redundancy and the potential for development of primary resistance to anti-cancer therapies. IGF-1R signaling can contribute to treatment resistance via general mechanisms that promote tumorigenesis, through crosstalk/redundancy with other RTK signaling networks that drive cellular proliferation and upregulation of anti-apoptotic proteins [49]. In addition, the IGF axis has specific effects relevant to individual classes of anti-cancer treatment. For example, IGF-1R signaling is thought to upregulate the activity of efflux pumps such as multidrug-resistance-associated protein 2, which can reduce the intracellular concentration of cytotoxic drugs [131]. IGF-1R signaling may also promote DSB repair following treatment with DNA-damaging agents, thereby reducing the efficacy of standard cytotoxic treatments [31, 32, 132]. Finally, crosstalk between the IGF axis and signaling via the ER [133] or AR [134] can promote treatment resistance by stimulating hormone-receptor-dependent transcriptional pathways.
In addition to primary treatment resistance, IGF signaling plays a key role in the development of acquired resistance to chemotherapeutic or targeted agents via feedback relief responses, which lead to compensatory activation of IGF survival signaling [135]. For example, acquired resistance to EGFR TKIs in lung squamous cell carcinoma and adenocarcinoma can result from IGF-1R activation, via the loss of the negative regulator IGFBP3 [135,136,137].
Given these mechanisms, IGF-targeted therapy lends itself as an attractive backbone to novel combination regimens as a strategy to prevent or overcome treatment resistance, and combinations that incorporate IGF-targeted agents with standard cytotoxic agents and/or other targeted therapies have been investigated preclinically in many human cancers (Table 3). The remainder of this review will summarize the current evidence to support various combination approaches and discuss potential opportunities for future clinical trials.
4.2 Combination with DNA-Damaging Agents
Preclinical evidence suggests that IGF signaling can protect tumor cells from chemotherapy- or radiotherapy-induced cell death [24, 31, 32, 138,139,140,141,142,143,144]. For example, in models of CRPC, upregulation of IGF-2 via the transcription factor GATA2 strongly contributed to chemotherapy resistance after docetaxel and cabazitaxel treatment, by activating numerous downstream kinases including IGF-1R and INSR. Moreover, dual IGF-1R/INSR inhibition restored the efficacy of both chemotherapy drugs and improved in vitro and in vivo survival [24]. In another study, exposure of murine glioma stem cells to fractionated radiation induced an increase in IGF-1 secretion and upregulation of IGF-1R, which together conferred resistance by protecting cells from subsequent radiation therapy [143].
Some studies support the importance of the scheduling of IGF-1R inhibition and chemotherapy, in order to achieve an optimal therapeutic response, suggesting that treatment sequencing approaches should be considered in future combination clinical trials incorporating these two treatment modalities [140, 141]. In contrast, combination with IGF-targeted therapies may increase radio-sensitivity without regard to scheduling of radiation and IGF-1R inhibition [142].
Inhibition of IGF-1R signaling is thought to enhance the effects of chemotherapy or radiotherapy primarily through suppression of DNA damage repair [5, 31, 32]. For example, addition of the anti-IGF-1R mAb R-1507 increased the anti-tumor activity of a standard cisplatin/ionizing radiation couplet in in vitro and in vivo models of SCLC. Moreover, genomic and functional analyses identified downregulation of the nucleotide excision repair pathway in SCLC cells treated with the R-1507/cis-IR combination [32].
With regard to clinical evidence to support the use of IGF-1R-targeted/cytotoxic drug combinations, IGF-1R overexpression in tumor tissue samples was found to be associated with shorter survival and a less favorable response to temozolomide chemotherapy in patients with glioblastoma multiforme (GBM) [167]. However, the results of combination trials with IGF-1R inhibitory drugs and chemotherapy have been disappointing. In a phase III trial in unselected patients with metastatic pancreatic cancer, a combination of ganitumab plus gemcitabine had manageable toxicity but did not improve OS compared with gemcitabine alone [123]. Similarly, addition of figitumumab to paclitaxel plus carboplatin failed to increase OS in patients with advanced nonadenocarcinoma NSCLC; this phase III study was closed early due to futility and an increased incidence of serious AEs and deaths in the figitumumab arm [168]. Furthermore, a phase II/III study of the IGF-1R mAb dalotuzumab in combination with cetuximab and irinotecan was discontinued prematurely due to lack of overall benefit with the addition of dalotuzumab, although there was a trend towards longer PFS and OS with this combination in patients with higher tumor IGF-1 mRNA [169]. Notably, none of these studies incorporated the sequential schedule of administration that was found to be optimal in preclinical studies [140, 141]. Based on these findings, and despite a convincing preclinical rationale, we suggest that factors contributing to failure of these clinical trials are likely to include toxicity, suboptimal treatment scheduling, and the lack of a predictive biomarker.
Regarding IGF-1R TKIs, three separate phase I/II clinical studies have evaluated the safety and efficacy of BMS-754807 in combination with paclitaxel plus carboplatin in patients with advanced solid tumors (NCT00793897), and with either trastuzumab (an HER-targeted mAb) or letrozole in patients with breast cancer (NCT00788333; NCT01225172); however, no results have been published, to-date. A phase II trial is investigating the efficacy and safety of istiratumab in combination with gemcitabine plus nab-paclitaxel in patients with metastatic pancreatic cancer, on the basis of preclinical evidence that istiratumab can induce significant tumor regression, including durable complete responses [57, 170].
4.3 Combination with Inhibitors of Other RTKs and Downstream Effectors
4.3.1 RTK Inhibitors
Crosstalk between the IGF axis and other RTK signaling pathways (including EGFR, HER2, VEGFR, PGDFR, cMET, and ALK) results in reciprocal compensatory mechanisms that limit response and/or mediate acquired resistance to treatments that target an individual pathway (Fig. 4) [49, 50, 63, 98,99,100,101,102,103,104,105,106,107, 137, 146, 171, 172].
This crosstalk is particularly apparent with regard to IGF and EGFR. In GBM, both of these pathways mediate activation of PI3K/Akt signaling [145]. Administration of AG1478, an EGFR TKI, resulted in IGF-1R upregulation in GBM cell lines, leading to sustained compensatory signaling through the PI3K pathway which produced potent anti-apoptotic and pro-invasion effects [145]. In another preclinical GBM study, the EGFR TKI gefitinib was effective in attenuating downstream MAPK/MEK/ERK activity, but concurrent inhibition of EGFR and IGF-1R/INSR was required to suppress Akt and induce apoptosis [105]. Similarly, in pediatric GBM, IGF-1R inhibition reduced signaling through the PI3K pathway, while targeting PDGFR resulted in parallel inhibition of signaling via the MAPK pathway. Targeting both receptors together resulted in enhanced anti-tumor activity compared with inhibition of either receptor alone [147].
Phospho-RTK array analysis in lung cancer models identified increased IGF-1R phosphorylation in erlotinib-resistant adenocarcinoma cells with wild-type EGFR [106]. In another study, NSCLC cell lines resistant to irreversible (e.g. PF299804) or mutant-selective (WZ4002) EGFR TKIs demonstrated activated IGF signaling due to loss of IGFBP3 expression, and combination of PF299804 or WZ4002 with the IGF-1R inhibitor BMS-536924 prevented the emergence of drug-resistant clones [137]. The sensitivity of NSCLC cells with acquired resistance to WZ4002 was also restored by AG-1024 (a small-molecule IGF-1R TKI) or xentuzumab [63]. IGF-1R knockdown sensitized resistant NSCLC cells to afatinib, another irreversible EGFR TKI, with combined treatment resulting in more than additive effects on tumor growth inhibition in vivo [103]. Inhibition of IGF-1R signaling using microRNAs has also been shown to partially reverse acquired resistance to erlotinib in NSCLC models, and sensitize HER2-positive breast cancer cells to HER-targeted drugs [100, 101]. Finally, IGF-2 overexpression has been observed in CRC patients whose tumors lacked resistance-conferring KRAS mutations but who responded to the EGFR mAb cetuximab with stable disease rather than an objective response. Furthermore, inhibition of IGF-1R/INSR signaling with BMS-754807 potentiated the anti-tumor effects of cetuximab in IGF2-overexpressing xenografts [107].
Based on this preclinical evidence with EGFR and IGF-1R inhibitors, together with the potential for better tolerability of IGF-ligand blockers, xentuzumab is under clinical investigation in combination with afatinib in a phase Ib trial in patients with NSCLC (NCT02191891). This combination demonstrated a manageable safety profile and encouraging activity; at the RP2D, one of 12 patients had a partial response and 10 patients had stable disease [173].
Activation of IGF-1R signaling has also been demonstrated in models of ALK TKI resistance, supporting the use of combined treatment with IGF-targeted therapies and ALK inhibitors, or second-generation ALK TKIs such as ceritinib (LDK-378), which simultaneously inhibits ALK and IGF-1R phosphorylation [104].
4.3.2 PI3K/Akt/mTOR and MAPK Pathway Inhibitors
With regard to downstream effector pathways, reciprocal interactions between Src and IGF-1R signaling have been shown to promote treatment resistance in prostate and lung cancer models; thus, combining IGF-targeted agents with drugs that target Src may be more effective than targeting either signaling component alone [148, 149]. Indeed, there is evidence that co-targeting IGF-1R and Src family kinases by combining anti-IGF-1R mAbs/IGF-1R TKIs with dasatinib resulted in more effective growth inhibition than single-agent treatments in rhabdomyosarcoma cell lines [150].
Extensive preclinical evidence suggests that IGF-1R activation can also mediate acquired resistance to inhibitors of the PI3K/Akt/mTOR and MAPK signaling pathways [22, 66, 74, 99, 151,152,153,154, 157, 174], not least because drugs that target individual components of mitogenic signaling pathways can relieve feedback inhibition of other signaling components. For example, Akt inhibition can lead, via activation of FOXO transcription factors, to reciprocal upregulation of IGF-1R, INSR, and other RTKs to reactivate the PI3K/Akt/mTOR pathway. In an equivalent process, inhibition of mTORC1 activates Akt and ERK signaling by blocking S6K-dependent feedback and activating IGF-1R and HER kinases [99]. Thus, combined inhibition of multiple mitogenic signaling pathways that are reciprocally reactivated by feedback relief may show enhanced anti-tumor activity. In one preclinical study, combination of xentuzumab with mTORC1 inhibitor rapamycin significantly enhanced its anti-tumor activity in Ewing sarcoma, and responses were associated with effective neutralization of circulating bioactive IGF-1/IGF-2 and with suppression of Akt activity in tumor tissue [66]. Also in Ewing sarcoma, dusigitumab in combination with rapamycin or mTORC1/2 kinase inhibitor AZD2014 was more effective than single-agent dusigitumab in inhibiting in vivo tumor growth [74]. Several studies support the use of triple therapies that incorporate IGF-targeted agents and mTOR inhibitors with other drugs. For example, this concept is supported by a study of dalotuzumab, the mTORC1 inhibitor ridaforolimus, and letrozole in hormone-sensitive breast cancer, in which the greatest anti-tumor effects were observed when all three drugs were combined [175]. In addition, a recent preclinical study of IGF-1R/mTOR inhibition in Ewing sarcoma identified several potential bypass pathways leading to resistance, including upregulation of IRS1, PI3K, and STAT3; these signaling proteins represent additional targets that could be incorporated into future studies of combined IGF-1R/mTOR inhibitors [176]. Indeed, promising data have been reported using the novel agent NT157, which inhibits both IGF-1R-IRS1/2 and STAT3 signaling in tumor and stromal cells, and has potent anti-tumor effects in preclinical models of melanoma and colorectal cancer [158, 159].
In one preclinical study that aimed to identify potential combination partners for MAPK pathway inhibitors, a large-scale short hairpin RNA screen revealed an adaptive response involving the IGF-1R/PI3Kα network in BRAF-mutated melanoma cell lines with phosphatase and tensin homolog (PTEN) loss of function. Co-inhibition of the MAPK pathway, PI3Kβ, and either PI3Kα or IGF-1R synergistically sustained PI3K signaling blockade, induced apoptosis, and suppressed in vivo tumor growth [157]. Simultaneous inhibition of the PI3K/Akt and MAPK signaling pathways has also demonstrated clinical activity in patients with some human cancers, albeit at the expense of increased toxicity [177, 178]. However, KRAS can directly activate the MEK/ERK and PI3K/Akt signaling pathways, which may underlie the resistance of KRAS mutant cancers to TKIs [151, 179, 180]. Ebi et al. showed that RTKs, particularly IGF-1R, regulated PI3K signaling in KRAS mutant cell lines, suggesting that targeting IGF-1R in combination with MAPK pathway inhibition could be an effective treatment strategy in KRAS mutant cancers [151].
4.3.3 Other Targeted Agents
Disruption of the CDK4/6 axis can contribute to unscheduled proliferation and tumorigenesis in a variety of cancers that show particular genomic features, such as mutations in CDKN2A [181]. For this reason, selective CDK4/6 inhibitors have been developed as potential anti-cancer therapies. In addition, emerging evidence from drug screening studies suggests that IGF- and CDK4/6-targeted agents can have synergistic anti-tumor effects in models of pancreatic cancer and soft tissue sarcoma by co-operatively suppressing Akt signaling [160, 161]. Combined inhibition of IGF-1R and poly(ADP-ribose) polymerase (PARP) may also improve clinical efficacy by interfering with DNA damage repair, specifically in cancers with defective homologous recombination (HR) deficiency, found in approximately 50% of breast and ovarian cancers [162]. Moreover, BMS-754807 has been shown to sensitize MCF7 breast cancer cells to inhibitors of ataxia telangiectasia-mutated and RAD3-related kinase (ATR), both of which mediate DNA damage repair, and to potentiate the effects of cisplatin in some cell lines [164].
Finally, INSR signaling can contribute to tumor proliferation and survival, primarily through the INSR-A isoform [182]. Indeed, insulin-driven proliferative signaling through INSR-A is reported to be a compensatory mechanism after endocrine therapy in breast cancer [183]. Drugs such as the anti-diabetic drug metformin that improve insulin sensitivity and lower insulin levels could counteract these effects; thus, such treatments represent potential combination partners for therapies targeting the IGF axis. In support of this hypothesis, metformin enhanced the anti-tumor effects of figitumumab in SCLC and NSCLC cell lines. As metformin has both hypoglycemic and anti-cancer effects, this treatment may also offset some of the adverse effects observed with anti-IGF-1R mAbs and IGF-1R TKIs [22, 163]. Clinical data from ongoing trials combining IGF-targeting drugs such as ganitumab with metformin (I-SPY 2 trial; NCT01042379) are eagerly awaited.
4.3.4 Clinical Combination Studies with Targeted Agents
Clinical studies to-date have demonstrated feasibility of several targeted combination approaches, with a focus on inhibitors of EGFR and mTOR. Most studies involving anti-IGF-1R mAbs have identified only known class-effect toxicities and no new relevant side effects [155, 184,185,186,187]. However, similar to monotherapy studies, the majority of clinical trials evaluating regimens that incorporate IGF-1R-targeted agents with other targeted therapies have, so far, shown limited clinical benefit. In a phase I trial in patients with advanced solid tumors, treatment with the IGF-1R/INSR TKI linsitinib in combination with erlotinib was well tolerated, with preliminary evidence of clinical activity, particularly in patients with NSCLC. Moreover, durable responses were observed in patients unlikely to respond to erlotinib monotherapy [188]. However, a phase II study of the same combination in patients with NSCLC harboring activating EGFR mutations was terminated early due to inferiority in the linsitinib arm, together with increased toxicity that necessitated erlotinib dose reductions and discontinuations [189]. Addition of ganitumab to the EGFR Ab panitumumab failed to improve clinical outcomes in a randomized phase Ib/II trial in patients with CRC [190].
Outcomes have been similarly negative in trials testing combined IGF-1R/mTOR inhibition. There was no evidence of clinical activity in a phase Ib study in patients with metastatic CRC who received linsitinib in combination with the mTORC1 inhibitor everolimus [191]. Treatment with cixutumumab plus temsirolimus, another mTORC1 inhibitor, produced no objective responses in a phase II trial in children and young adults with refractory sarcoma [192], even though, in an earlier phase I study in adult cancer patients, this treatment induced objective responses in two of three patients with Ewing sarcoma and four of 10 patients with adrenocortical carcinoma [186]. In a phase I study in patients with neuroendocrine tumors, cixutumumab was assessed in combination with everolimus plus octreotide, a long-acting somatostatin analog, but was associated with adverse effects, including hyperglycemia, that may limit its long-term tolerance [193]. Recruitment to a phase II trial of dalotuzumab plus ridaforolimus in patients with ER-positive breast cancer was suspended due to a higher than expected rate of stomatitis in the combination arm, although the combination was shown to be tolerable with lower doses of ridaforolimus [194]. In a phase I trial in 87 patients with advanced cancers, the same combination resulted in six confirmed partial responses, including three patients with breast cancer. In total, 10 of 23 patients with breast cancer and six of 11 patients with ER-positive/high-proliferative breast cancer showed evidence of anti-tumor activity, based on objective responses, PFS, and tumor metabolic rates/biomarkers [156].
So far, clinical benefit with targeted combination therapies has been observed in only a minority of cases, which re-emphasizes the importance of better selection of patients who are most likely to benefit from combinatorial IGF-targeted therapy. This process could be aided by future research into the identification of biomarkers for IGF therapy response.
4.4 Combination with Endocrine Therapies
Interactions identified between the IGF axis and endocrine pathways provide a mechanistic rationale for investigating IGF-targeted therapies in combination with hormone therapy in breast and prostate cancers [133, 134, 164, 195,196,197]. In preclinical breast cancer models, inhibition of IGF-1R signaling with BMS-754807 was shown to overcome resistance to tamoxifen, a partial estrogen agonist, and letrozole, a non-steroidal aromatase inhibitor (AI), in ER-positive MCF-7 cells [133]. In prostate cancer, androgen stimulation induced IGF-1 upregulation in AR-positive cells and enhanced their proliferation and invasiveness [196]. Furthermore, inhibition of IGF-1R potentiated regression of prostate cancer xenografts and delayed recurrence following castration [134]. Of note, different AR splice variants may differentially alter IGF-axis components, as stimulation of full-length AR (ARfl) or ARv567es (a constitutively active variant that skips exons 5, 6, and 7) has been shown to enhance or suppress IGF-1R expression, respectively [198]. In PTEN-mutated prostate cancer, inhibition of PI3Kβ relieved feedback inhibition of IGF-1R and other RTKs, leading to activation of PI3Kα and downstream signaling pathways. Combined inhibition of PI3Kα and PI3Kβ suppressed this feedback relief, but resulted in potent AR activation; therefore, combined inhibition of both PI3K isoforms and AR may be required to maximize tumor regression [174]. In studies performed to better understand the molecular determinants of IGF-1R overexpression in prostate cancer, expression of TMPRSS2-ERG (T2E), an androgen-regulated oncogenic fusion protein, was shown to upregulate IGF-1R mRNA and protein expression, whilst overexpression of truncated ERG increased the sensitivity of prostate cancer cells to the IGF-1R TKI NVP-AEW541 and to anti-IGF-1R mAbs figitumumab and AVE1642 [165, 199]. Furthermore, co-administration of figitumumab and abiraterone, an irreversible inhibitor of cytochrome P450 17A1 that prevents androgen synthesis, resulted in synergistic anti-proliferative activity in T2E-expressing cell lines [165].
Despite the preclinical evidence supporting evaluation of IGF-targeted and endocrine therapy combinations, treatment with ganitumab plus the AI exemestane or ER antagonist fulvestrant did not improve PFS in women with previously treated hormone receptor-positive breast cancer, and OS was lower in the combination group versus endocrine treatment alone [197, 200]. Furthermore, planned biomarker analyses, including PI3KCA mutations, could not identify any patient subgroups that benefited from ganitumab [197]. The IGF-1R TKI KW-2450 has shown modest single-agent clinical activity in phase I, with dose-limiting toxicities of hyperglycemia and rash [62]. However, another phase I study of this agent, in combination with lapatinib and letrozole in HER2-positive breast cancer (NCT01199367), was terminated and no results have been made available.
Several current studies are testing IGF ligand-neutralizing Abs with endocrine therapy. Dusigitumab is in phase II development in combination with an AI, for the first-line treatment of metastatic breast cancer (NCT01446159); however, data are not yet available. A phase Ib/II trial is also investigating the safety and efficacy of xentuzumab in combination with exemestane and everolimus in the same setting. At the RP2D (xentuzumab 1000 mg/week plus exemestane 25 mg/day and everolimus 10 mg/day), this triple combination had a manageable safety profile and encouraging preliminary anti-tumor activity, with 62% of patients achieving disease control and 24% of patients achieving a partial response [201].
In a prospective neoadjuvant study in prostate cancer, treatment with cixutumumab in combination with androgen-deprivation therapy (ADT) resulted in significant changes in serum components of the IGF and glucose homeostasis pathways (growth hormone, IGF-1, IGF-2, IGFBP3, and insulin) versus ADT alone, albeit only in overweight or obese patients. This finding further highlights the need for appropriate patient selection in clinical trials of IGF-targeted therapies [195]. For patients with CRPC, clinical practice guidelines recommend the use of chemotherapy and/or targeted agents in addition to continued hormone therapy. These treatments, examples of which include abiraterone and enzalutamide (an AR antagonist) [202, 203], could also be considered as combination partners for IGF-targeted therapies. A phase Ib/II study is currently recruiting patients with metastatic CRPC following progression on docetaxel, to evaluate treatment with xentuzumab in combination with enzalutamide, versus enzalutamide alone (NCT02204072).
4.5 Combination with Immune Therapy
Abs that inhibit immune checkpoints, particularly those directed against cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed death 1 (PD-1), and programmed death ligand 1, have shown very encouraging efficacy in melanoma and NSCLC [204,205,206,207,208,209]. However, although immunotherapies are associated with durable tumor control, objective response rates are relatively low. In an attempt to improve response rates, combination regimens that incorporate both targeted agents and immunotherapies are currently undergoing clinical evaluation in a wide range of cancers [210].
Several lines of evidence provide a rationale for combining IGF-signaling inhibitors and immune therapies. The contribution of the IGF axis in regulating immune function was first highlighted in a series of studies reporting that downregulating IGF-1 or IGF-1R enhanced the immunogenicity of GBM models in rats, as well as in a small clinical study in astrocytoma [211,212,213]. Subsequent studies revealed that IGF-1 alleviates a number of autoimmune conditions, including autoimmune diabetes and contact dermatitis, and that its protective effect appears to be correlated with increased infiltration by regulatory T cells in affected tissues [214,215,216]. IGF-1 has also been implicated in the expression of immunosuppressive cytokines including interleukin-10 [217, 218], as well as induction of the M2 phenotype of macrophages [219]. In preclinical models of HCC, expression of IGF-1 by M2 macrophages increased hepatoma growth. Furthermore, sorafenib treatment reduced macrophage IGF-1 expression and IGF-1-driven cell proliferation, associated with altered macrophage polarization [220].
A recent study in pancreatic ductal adenocarcinoma suggested a role for IGF signaling in tumor infiltration of tumor-associated macrophages (TAMs). A positive correlation was identified between activated insulin and IGF receptors, which were expressed in tumor biopsies from 72% of patients, and infiltration of TAMs. Furthermore, the data indicated that TAMs and tumor stromal myofibroblasts can directly support chemo-resistance, by secreting IGF-1 and IGF-2 [139]. In addition to these findings, inhibition of IGF-1/IGF-2 with xentuzumab sensitized tumors to gemcitabine in a murine pancreatic cancer model [139]. Activation of IGF-1R signaling through stimulation by TAM-derived IGF-1 has also been shown to mediate acquired resistance to colony stimulating factor 1 receptor (CSF-1R) inhibitors in glioma. Moreover, in genetic mouse models of GBM, combined CSF-1R and IGF-1R inhibition significantly improved survival outcomes [166].
Finally, cell death induced by IGF-1R inhibition may lead to the release of tumor-specific antigens (TSAs) that help to initiate and potentiate immune responses facilitated by immunotherapeutic agents. Compared with circulating B cells, tumor-infiltrating B cells in melanoma patients show increased expression of inflammatory cytokines and growth factors, including IGF-1, which could reduce TSA release within the tumor microenvironment, potentially leading to resistance to immune checkpoint inhibitors such as anti-CTLA-4 and anti-PD-1 mAbs [221].
5 Conclusions and Future Perspectives
Dysregulation of the IGF axis can contribute to many of the hallmarks of cancer and is implicated in the development of resistance to a number of standard-of-care therapies. Thus, the IGF axis is an attractive therapeutic target. Despite this, the lack of a bona fide predictive biomarker for patient enrichment has impeded the clinical development of IGF-targeted therapies. Pre-treatment circulating or tumor IGF levels have been proposed as a potential candidate by several groups [116,117,118,119,120,121,122,123, 125, 169], although such hypotheses will need to be tested and confirmed in randomized, stratified clinical trials. There is clearly meaningful activity and long-term benefit from IGF-targeted monotherapies in patients with Ewing sarcoma and soft-tissue sarcoma [81,82,83]. In common cancers, the key to success will be to develop mechanistically rational combination therapies, reflecting the role of the IGF axis as an epi-driver and bypass pathway [52, 80, 110].
Given that IGF-neutralizing Abs inhibit signaling through both IGF-1R and INSR-A, without impacting insulin and glucose metabolism [66, 75], combination of these agents with other types of anti-cancer treatment could represent a more clinically effective and tolerable approach compared with combinations that include IGF-1R mAbs or TKIs. Based on preclinical and clinical evidence to date, we suggest that the key priorities for future studies should focus on the following combination approaches/partners for IGF ligand-neutralizing Abs: (1) EGFR family inhibitors, such as afatinib, particularly in the context of resistance to third-generation EGFR TKIs; (2) CDK inhibitors, given that IGF-1R has been identified in screening studies as having a role in mediating resistance to these drugs; (3) endocrine therapy, since sensitivity to IGF-targeted agents has been observed in ER-positive breast cancer models; (4) immune checkpoint inhibitors, based on the roles of IGF ligands in promoting an immunosuppressive environment; and (5) radiotherapy, although no clinical trial has tested this hypothesis yet, despite the supportive preclinical data.
Future clinical research of IGF-targeted therapies may also be improved by incorporating Adaptive Design Clinical Trials, which include prospectively planned opportunities for modification of the study design based on analysis during the study [222]. Coupled with precise measurement of IGF ligands in the circulation, and IGFs and receptors in clinical tumor tissue, this approach could potentially reveal effective combination regimens and help to identify predictive biomarkers for better selection of patients most likely to derive clinical benefit.
References
Brahmkhatri VP, Prasanna C, Atreya HS. Insulin-like growth factor system in cancer: novel targeted therapies. Biomed Res Int. 2015;2015:538019. doi:10.1155/2015/538019.
Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015;2(1):13–25. doi:10.1016/j.gendis.2014.10.004.
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12(3):159–69. doi:10.1038/nrc3215.
Evdokimova V, Tognon CE, Benatar T, Yang W, Krutikov K, Pollak M, et al. IGFBP7 binds to the IGF-1 receptor and blocks its activation by insulin-like growth factors. Sci Signal. 2012;5(255):ra92. doi:10.1126/scisignal.2003184.
Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res. 2008;14(20):6364–70. doi:10.1158/1078-0432.CCR-07-4879.
Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986;5(10):2503–12.
Li R, Pourpak A, Morris SW. Inhibition of the insulin-like growth factor-1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J Med Chem. 2009;52(16):4981–5004. doi:10.1021/jm9002395.
Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer. 2014;14(5):329–41. doi:10.1038/nrc3720.
Lin MZ, Marzec KA, Martin JL, Baxter RC. The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents. Oncogene. 2014;33(1):85–96. doi:10.1038/onc.2012.538.
Jin M, Buck E, Mulvihill MJ. Modulation of insulin-like growth factor-1 receptor and its signaling network for the treatment of cancer: current status and future perspectives. Oncol Rev. 2013;7(1):e3. doi:10.4081/oncol.2013.e3.
Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6(1):1–12. doi:10.1158/1535-7163.MCT-06-0080.
van de Luijtgaarden AC, Versleijen-Jonkers YM, Roeffen MH, Schreuder HW, Flucke UE, van der Graaf WT. Prognostic and therapeutic relevance of the IGF pathway in Ewing’s sarcoma patients. Target Oncol. 2013;8(4):253–60. doi:10.1007/s11523-012-0248-3.
Sell C, Dumenil G, Deveaud C, Miura M, Coppola D, DeAngelis T, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994;14(6):3604–12.
Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer. 2006;13(suppl 1):S33–43. doi:10.1677/erc.1.01280.
Werner H, Roberts CT Jr. The IGFI receptor gene: a molecular target for disrupted transcription factors. Genes Chromosom Cancer. 2003;36(2):113–20. doi:10.1002/gcc.10157.
Werner H, Roberts CT Jr, Rauscher FJ 3rd, LeRoith D. Regulation of insulin-like growth factor I receptor gene expression by the Wilms’ tumor suppressor WT1. J Mol Neurosci. 1996;7(2):111–23.
Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13. doi:10.1126/scitranslmed.3001845.
Steuerman R, Shevah O, Laron Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur J Endocrinol. 2011;164(4):485–9. doi:10.1530/EJE-10-0859.
Burrow S, Andrulis IL, Pollak M, Bell RS. Expression of insulin-like growth factor receptor, IGF-1, and IGF-2 in primary and metastatic osteosarcoma. J Surg Oncol. 1998;69(1):21–7.
Steller MA, Delgado CH, Bartels CJ, Woodworth CD, Zou Z. Overexpression of the insulin-like growth factor-1 receptor and autocrine stimulation in human cervical cancer cells. Cancer Res. 1996;56(8):1761–5.
King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev. 2014;40(9):1096–105. doi:10.1016/j.ctrv.2014.07.004.
Cao H, Dong W, Shen H, Xu J, Zhu L, Liu Q, et al. Combinational therapy enhances the effects of Anti-IGF-1R mAb Figitumumab to target small cell lung cancer. PLoS One. 2015;10(8):e0135844. doi:10.1371/journal.pone.0135844.
Livingstone C. IGF2 and cancer. Endocr Relat Cancer. 2013;20(6):R321–39. doi:10.1530/ERC-13-0231.
Vidal SJ, Rodriguez-Bravo V, Quinn SA, Rodriguez-Barrueco R, Lujambio A, Williams E, et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell. 2015;27(2):223–39. doi:10.1016/j.ccell.2014.11.013.
Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther. 2010;9(10):2652–64. doi:10.1158/1535-7163.MCT-10-0318.
Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 2007;67(1):391–7. doi:10.1158/0008-5472.CAN-06-1712.
Martinez-Quetglas I, Pinyol R, Dauch D, Torrecilla S, Tovar V, Moeini A, et al. IGF2 is upregulated by epigenetic mechanisms in hepatocellular carcinomas and is an actionable oncogene product in experimental models. Gastroenterology. 2016;151(6):1192–1205 doi:10.1053/j.gastro.2016.09.001.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi:10.1016/j.cell.2011.02.013.
Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne). 2014;5:10. doi:10.3389/fendo.2014.00010.
Heerboth S, Housman G, Leary M, Longacre M, Byler S, Lapinska K, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4:6. doi:10.1186/s40169-015-0048-3.
Chitnis MM, Lodhia KA, Aleksic T, Gao S, Protheroe AS, Macaulay VM. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene. 2014;33(45):5262–73. doi:10.1038/onc.2013.460.
Ferte C, Loriot Y, Clemenson C, Commo F, Gombos A, Bibault JE, et al. IGF-1R targeting increases the antitumor effects of DNA-damaging agents in SCLC model: an opportunity to increase the efficacy of standard therapy. Mol Cancer Ther. 2013;12(7):1213–22. doi:10.1158/1535-7163.MCT-12-1067.
Turney BW, Kerr M, Chitnis MM, Lodhia K, Wang Y, Riedemann J, et al. Depletion of the type 1 IGF receptor delays repair of radiation-induced DNA double strand breaks. Radiother Oncol. 2012;103(3):402–9. doi:10.1016/j.radonc.2012.03.009.
Bowers LW, Rossi EL, O’Flanagan CH, de Graffenried LA, Hursting SD. The role of the insulin/IGF system in cancer: lessons learned from clinical trials and the energy balance-cancer link. Front Endocrinol (Lausanne). 2015;6:77. doi:10.3389/fendo.2015.00077.
Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65(6):1053–61.
Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7(3):331–42.
Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A. 1995;92(26):12146–50.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–9. doi:10.1038/378785a0.
Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8(10):1145–52. doi:10.1038/nm759.
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3(3):245–52. doi:10.1038/35060032.
Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12(4):487–502. doi: 10.1016/j.devcel.2007.03.020.
Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;14(27(8)):1106–13. doi: 10.1038/sj.onc.1210715.
Aleksic T, Chitnis MM, Perestenko OV, Gao S, Thomas PH, Turner GD, et al. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010;70(16):6412–9. doi:10.1158/0008-5472.CAN-10-0052.
Deng H, Lin Y, Badin M, Vasilcanu D, Stromberg T, Jernberg-Wiklund H, et al. Over-accumulation of nuclear IGF-1 receptor in tumor cells requires elevated expression of the receptor and the SUMO-conjugating enzyme Ubc9. Biochem Biophys Res Commun. 2011;404(2):667–71. doi:10.1016/j.bbrc.2010.12.038.
Sarfstein R, Werner H. Minireview: nuclear insulin and insulin-like growth factor-1 receptors: a novel paradigm in signal transduction. Endocrinology. 2013;154(5):1672–9. doi:10.1210/en.2012-2165.
Sehat B, Tofigh A, Lin Y, Trocme E, Liljedahl U, Lagergren J, et al. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci Signal. 2010;3(108):ra10. doi:10.1126/scisignal.2000628.
Sarfstein R, Belfiore A, Werner H. Identification of insulin-like growth factor-I receptor (IGF-IR) Gene promoter-binding proteins in estrogen receptor (ER)-positive and ER-depleted breast cancer cells. Cancers (Basel). 2010;2(2):233–61. doi:10.3390/cancers2020233.
Girnita L, Worrall C, Takahashi S, Seregard S, Girnita A. Something old, something new and something borrowed: emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol Life Sci. 2014;71(13):2403–27. doi:10.1007/s00018-013-1514-y.
Liu C, Zhang Z, Tang H, Jiang Z, You L, Liao Y. Crosstalk between IGF-1R and other tumor promoting pathways. Curr Pharm Des. 2014;20(17):2912–21.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–9. doi:10.1038/nature11249.
Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front Pharmacol. 2013;4:30. doi:10.3389/fphar.2013.00030.
Craddock BP, Miller WT. Effects of somatic mutations in the C-terminus of insulin-like growth factor 1 receptor on activity and signaling. J Signal Transduct. 2012;2012:804801. doi:10.1155/2012/804801.
Beltran PJ, Mitchell P, Chung YA, Cajulis E, Lu J, Belmontes B, et al. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol Cancer Ther. 2009;8(5):1095–1105. doi:10.1158/1535-7163.MCT-08-1171.
Zhang T, Shen H, Dong W, Qu X, Liu Q, Du J. Antitumor effects and molecular mechanisms of figitumumab, a humanized monoclonal antibody to IGF-1 receptor, in esophageal carcinoma. Sci Rep. 2014;4:6855. doi:10.1038/srep06855.
Kurzrock R, Patnaik A, Aisner J, Warren T, Leong S, Benjamin R, et al. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin Cancer Res. 2010;16(8):2458–65. doi:10.1158/1078-0432.CCR-09-3220.
Wang Y, Hailey J, Williams D, Wang Y, Lipari P, Malkowski M, et al. Inhibition of insulin-like growth factor-I receptor (IGF-IR) signaling and tumor cell growth by a fully human neutralizing anti-IGF-IR antibody. Mol Cancer Ther. 2005;4(8):1214–21. doi:10.1158/1535-7163.MCT-05-0048.
Fitzgerald JB, Johnson BW, Baum J, Adams S, Iadevaia S, Tang J, et al. MM-141, an IGF-IR- and ErbB3-directed bispecific antibody, overcomes network adaptations that limit activity of IGF-IR inhibitors. Mol Cancer Ther. 2014;13(2):410–25. doi:10.1158/1535-7163.MCT-13-0255.
Reidy-Lagunes DL, Vakiani E, Segal MF, Hollywood EM, Tang LH, Solit DB, et al. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer. 2012;118(19):4795–800. doi:10.1002/cncr.27459.
Abou-Alfa GK, Capanu M, O’Reilly EM, Ma J, Chou JF, Gansukh B, et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J Hepatol. 2014;60(2):319–24. doi:10.1016/j.jhep.2013.09.008.
Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8(12):3341–9. doi:10.1158/1535-7163.MCT-09-0499.
Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, et al. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem. 2009;1(6):1153–71. doi:10.4155/fmc.09.89.
Schwartz GK, Dickson MA, LoRusso PM, Sausville EA, Maekawa Y, Watanabe Y, et al. Preclinical and first-in-human phase I studies of KW-2450, an oral tyrosine kinase inhibitor with insulin-like growth factor receptor-1/insulin receptor selectivity. Cancer Sci. 2016;107(4):499–506. doi:10.1111/cas.12906.
Park JH, Choi YJ, Kim SY, Lee JE, Sung KJ, Park S, et al. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget. 2016;7(16):22005–15. doi:10.18632/oncotarget.8013.
Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, et al. In vivo antitumor activity of NVP-AEW541-a novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5(3):231–9.
Gao J, Chesebrough JW, Cartlidge SA, Ricketts SA, Incognito L, Veldman-Jones M, et al. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res. 2011;71(3):1029–40. doi:10.1158/0008-5472.CAN-10-2274.
Friedbichler K, Hofmann MH, Kroez M, Ostermann E, Lamche HR, Koessl C, et al. Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand-neutralizing antibody, and mechanistic rationale for combination with rapamycin. Mol Cancer Ther. 2014;13(2):399–409. doi:10.1158/1535-7163.MCT-13-0598.
Pandini G, Wurch T, Akla B, Corvaia N, Belfiore A, Goetsch L. Functional responses and in vivo anti-tumour activity of h7C10: a humanised monoclonal antibody with neutralising activity against the insulin-like growth factor-1 (IGF-1) receptor and insulin/IGF-1 hybrid receptors. Eur J Cancer. 2007;43(8):1318–27. doi:10.1016/j.ejca.2007.03.009.
Sachdev D, Singh R, Fujita-Yamaguchi Y, Yee D. Down-regulation of insulin receptor by antibodies against the type I insulin-like growth factor receptor: implications for anti-insulin-like growth factor therapy in breast cancer. Cancer Res. 2006;66(4):2391–402. doi:10.1158/0008-5472.CAN-05-3126.
Feng Y, Dimitrov DS. Antibody-based therapeutics against components of the IGF system. Oncoimmunology. 2012;1(8):1390–1. doi:10.4161/onci.20925.
De Meyts P, Palsgaard J, Sajid W, Theede AM, Aladdin H. Structural biology of insulin and IGF-1 receptors. Novartis Found Symp. 2004;262:160–71. discussion 71-6, 265-8.
Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodriguez-Braun E, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17(19):6304–12. doi:10.1158/1078-0432.CCR-10-3336.
Pollak M. Insulin, insulin-like growth factors and neoplasia. Best Pract Res Clin Endocrinol Metab. 2008;22(4):625–38. doi:10.1016/j.beem.2008.08.004.
Weroha SJ, Haluska P. IGF-1 receptor inhibitors in clinical trials--early lessons. J Mammary Gland Biol Neoplasia. 2008;13(4):471–83. doi:10.1007/s10911-008-9104-6.
Zhong H, Fazenbaker C, Breen S, Chen C, Huang J, Morehouse C, et al. MEDI-573, alone or in combination with mammalian target of rapamycin inhibitors, targets the insulin-like growth factor pathway in sarcomas. Mol Cancer Ther. 2014;13(11):2662–73. doi:10.1158/1535-7163.MCT-14-0144.
Haluska P, Menefee M, Plimack ER, Rosenberg J, Northfelt D, LaVallee T, et al. Phase I dose-escalation study of MEDI-573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(18):4747–57. doi:10.1158/1078-0432.CCR-14-0114.
Iguchi H, Nishina T, Nogami N, Kozuki T, Yamagiwa Y, Yagawa K. Phase I dose-escalation study evaluating safety, tolerability and pharmacokinetics of MEDI-573, a dual IGF-I/II neutralizing antibody, in Japanese patients with advanced solid tumours. Investig New Drugs. 2015;33(1):194–200. doi:10.1007/s10637-014-0170-x.
Rihawi K, Ong M, Michalarea V, Bent L, Buschke S, Bogenrieder T et al. Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumors. J Clin Oncol. 2014;32(5 suppl):abstr 2622.
Lin CC, Chang KY, Huang DC, Marriott V, Beijsterveldt LV, Chen LT, et al. A phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers. J Clin Oncol. 2014;32(5 suppl):abstr 2617.
Doi T, Shitara K, Naito Y, Kuboki Y, Kojima T, Hosono A, et al. Phase I dose escalation trial of weekly intravenous xentuzumab (BI 836845) in Japanese patients with advanced solid tumors. Ann Oncol. 2016;27(suppl):abstr 2790.
Baserga R. The decline and fall of the IGF-I receptor. J Cell Physiol. 2013;228(4):675–9. doi:10.1002/jcp.24217.
Malempati S, Weigel B, Ingle AM, Ahern CH, Carroll JM, Roberts CT, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology group. J Clin Oncol. 2012;30(3):256–62. doi:10.1200/JCO.2011.37.4355.
Schoffski P, Adkins D, Blay JY, Gil T, Elias AD, Rutkowski P, et al. An open-label, phase 2 study evaluating the efficacy and safety of the anti-IGF-1R antibody cixutumumab in patients with previously treated advanced or metastatic soft-tissue sarcoma or Ewing family of tumours. Eur J Cancer. 2013;49(15):3219–28. doi:10.1016/j.ejca.2013.06.010.
Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11(2):129–35. doi:10.1016/S1470-2045(09)70354-7.
Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol. 2009;27(34):5800–7. doi:10.1200/JCO.2009.23.6745.
Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a sarcoma alliance for research through collaboration study. Cancer. 2014;120(16):2448–56. doi:10.1002/cncr.28728.
Chiappori AA, Otterson GA, Dowlati A, Traynor AM, Horn L, Owonikoko TK, et al. A randomized phase II study of Linsitinib (OSI-906) versus Topotecan in patients with relapsed small-cell lung cancer. Oncologist. 2016;21(10):1163–4. doi:10.1634/theoncologist.2016-0220.
Schmitz S, Kaminsky-Forrett MC, Henry S, Zanetta S, Geoffrois L, Bompas E, et al. Phase II study of figitumumab in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: clinical activity and molecular response (GORTEC 2008-02). Ann Oncol. 2012;23(8):2153–61. doi:10.1093/annonc/mdr574.
Glisson BS, Tseng J, Marur S, Shin DM, Murphy BA, Cohen EEW et al. Randomized phase II trial of cixutumumab (CIX) alone or with cetuximab (CET) for refractory recurrent/metastatic squamous cancer of head and neck (R/M-SCCHN). J Clin Oncol. 2013;31(suppl):abstr 6030.
Isakoff SJ, Saleh MN, Lugovskoy AA, Mathews S, Czibere AG, Shields AF et al. First-in-human study of MM-141: a novel tetravalent monoclonal antibody targeting IGF-1R and ErbB3. J Clin Oncol. 2015;33(suppl 3):abstr 384.
Becerra CR, Salazar R, Garcia-Carbonero R, Thomas AL, Vazquez-Mazon FJ, Cassidy J, et al. Figitumumab in patients with refractory metastatic colorectal cancer previously treated with standard therapies: a nonrandomized, open-label, phase II trial. Cancer Chemother Pharmacol. 2014;73(4):695–702. doi:10.1007/s00280-014-2391-2.
Haluska P, Worden F, Olmos D, Yin D, Schteingart D, Batzel GN, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol. 2010;65(4):765–73. doi:10.1007/s00280-009-1083-9.
Gradishar WJ, Yardley DA, Layman R, Sparano JA, Chuang E, Northfelt DW, et al. Clinical and translational results of a phase II, randomized trial of an Anti-IGF-1R (Cixutumumab) in women with breast cancer that progressed on endocrine therapy. Clin Cancer Res. 2016;22(2):301–9. doi:10.1158/1078-0432.CCR-15-0588.
Ray-Coquard I, Haluska P, O’Reilly S, Cottu PH, Elit L, Provencher DM, et al. A multicenter open-label phase II study of the efficacy and safety of ganitumab (AMG 479), a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R) as second-line therapy in patients with recurrent platinum-sensitive ovarian cancer. J Clin Oncol. 2013;31(suppl):abstr 5515.
Desai J, Solomon BJ, Davis ID, Lipton LR, Hicks R, Scott AM, et al. Phase I dose-escalation study of daily BMS-754807, an oral, dual IGF-1R/insulin receptor (IR) inhibitor in subjects with solid tumors. J Clin Oncol. 2010;28(suppl 15):abstr 3104.
Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13(19):5834–40. doi:10.1158/1078-0432.CCR-07-1118.
Jones RL, Kim ES, Nava-Parada P, Alam S, Johnson FM, Stephens AW, et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin Cancer Res. 2015;21(4):693–700. doi:10.1158/1078-0432.CCR-14-0265.
Anderson PM, Bielack SS, Gorlick RG, Skubitz K, Daw NC, Herzog CE, et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr Blood Cancer. 2016;63(10):1761–70. doi:10.1002/pbc.26087.
Browne BC, Crown J, Venkatesan N, Duffy MJ, Clynes M, Slamon D, et al. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann Oncol. 2011;22(1):68–73. doi:10.1093/annonc/mdq349.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. doi:10.1016/j.ccr.2010.10.031.
Corcoran C, Rani S, Breslin S, Gogarty M, Ghobrial IM, Crown J, et al. miR-630 targets IGF1R to regulate response to HER-targeting drugs and overall cancer cell progression in HER2 over-expressing breast cancer. Mol Cancer. 2014;13:71. doi:10.1186/1476-4598-13-71.
Han J, Zhao F, Zhang J, Zhu H, Ma H, Li X, et al. miR-223 reverses the resistance of EGFR-TKIs through IGF1R/PI3K/Akt signaling pathway. Int J Oncol. 2016;48(5):1855–67. doi:10.3892/ijo.2016.3401.
Ioannou N, Seddon AM, Dalgleish A, Mackintosh D, Modjtahedi H. Treatment with a combination of the ErbB (HER) family blocker afatinib and the IGF-IR inhibitor, NVP-AEW541 induces synergistic growth inhibition of human pancreatic cancer cells. BMC Cancer. 2013;13(1):1–12. doi:10.1186/1471-2407-13-41.
Lee Y, Wang Y, James M, Jeong JH, You M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol Carcinog. 2016;55(5):991–1001. doi:10.1002/mc.22342.
Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20(9):1027–34. doi:10.1038/nm.3667.
Ma Y, Tang N, Thompson RC, Mobley BC, Clark SW, Sarkaria JN, et al. InsR/IGF1R pathway mediates resistance to EGFR inhibitors in glioblastoma. Clin Cancer Res. 2016;22(7):1767–76. doi:10.1158/1078-0432.CCR-15-1677.
Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int J Cancer. 2014;135(4):1002–6. doi:10.1002/ijc.28737.
Zanella ER, Galimi F, Sassi F, Migliardi G, Cottino F, Leto SM, et al. IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti-EGFR therapies. Sci Transl Med. 2015;7(272):272ra12. doi:10.1126/scitranslmed.3010445.
Rota LM, Wood TL. Crosstalk of the insulin-like growth factor receptor with the Wnt signaling pathway in breast cancer. Front Endocrinol (Lausanne). 2015;6:92. doi:10.3389/fendo.2015.00092.
Rota LM, Albanito L, Shin ME, Goyeneche CL, Shushanov S, Gallagher EJ, et al. IGF1R inhibition in mammary epithelia promotes canonical Wnt signaling and Wnt1-driven tumors. Cancer Res. 2014;74(19):5668–79. doi:10.1158/0008-5472.CAN-14-0970.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi:10.1126/science.1235122.
Hoyo C, Schildkraut JM, Murphy SK, Chow WH, Vaughan TL, Risch H, et al. IGF2R polymorphisms and risk of esophageal and gastric adenocarcinomas. Int J Cancer. 2009;125(11):2673–8. doi:10.1002/ijc.24623.
Oh SY, Shin A, Kim SG, Hwang JA, Hong SH, Lee YS, et al. Relationship between insulin-like growth factor axis gene polymorphisms and clinical outcome in advanced gastric cancer patients treated with FOLFOX. Oncotarget. 2016;7(21):31204–14. doi:10.18632/oncotarget.9100.
Zhang M, Hu Z, Huang J, Shu Y, Dai J, Jin G, et al. A 3′-untranslated region polymorphism in IGF1 predicts survival of non-small cell lung cancer in a Chinese population. Clin Cancer Res. 2010;16(4):1236–44. doi:10.1158/1078-0432.CCR-09-2719.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi:10.1038/nature08822.
Dziadziuszko R, Merrick DT, Witta SE, Mendoza AD, Szostakiewicz B, Szymanowska A, et al. Insulin-like growth factor receptor 1 (IGF1R) gene copy number is associated with survival in operable non-small-cell lung cancer: a comparison between IGF1R fluorescent in situ hybridization, protein expression, and mRNA expression. J Clin Oncol. 2010;28(13):2174–80. doi:10.1200/JCO.2009.24.6611.
Juergens H, Daw NC, Geoerger B, Ferrari S, Villarroel M, Aerts I, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–40. doi:10.1200/JCO.2010.33.0670.
Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II sarcoma alliance for research through collaboration study. J Clin Oncol. 2011;29(34):4541–7. doi:10.1200/JCO.2010.34.0000.
Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–56. doi:10.1200/JCO.2011.37.2359.
Goto Y, Sekine I, Tanioka M, Shibata T, Tanai C, Asahina H, et al. Figitumumab combined with carboplatin and paclitaxel in treatment-naive Japanese patients with advanced non-small cell lung cancer. Investig New Drugs. 2012;30(4):1548–56. doi:10.1007/s10637-011-9715-4.
Weickhardt A, Doebele R, Oton A, Lettieri J, Maxson D, Reynolds M, et al. A phase I/II study of erlotinib in combination with the anti-insulin-like growth factor-1 receptor monoclonal antibody IMC-A12 (cixutumumab) in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2012;7(2):419–26. doi:10.1097/JTO.0b013e31823c5b11.
Jalve MM, Shroff RT, Varadhachary GR, Wolff RA, Fogelman DR, Bhosale P, et al. Tumor IGF-1 expression as a predictive biomarker for IGF1R-directed therapy in advanced pancreatic cancer (APC). J Clin Oncol. 2012;30(suppl):abstr 4054.
McCaffery I, Tudor Y, Deng H, Tang R, Suzuki S, Badola S, et al. Putative predictive biomarkers of survival in patients with metastatic pancreatic adenocarcinoma treated with gemcitabine and ganitumab, an IGF1R inhibitor. Clin Cancer Res. 2013;19(15):4282–9. doi:10.1158/1078-0432.CCR-12-1840.
Fuchs CS, Azevedo S, Okusaka T, Van Laethem JL, Lipton LR, Riess H, et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trial. Ann Oncol. 2015;26(5):921–7. doi:10.1093/annonc/mdv027.
Lugovskoy AA, Curley M, Baum J, Adams S, Iadevaia S, Rimkunas V, et al. Preclinical characterization and first-in-human study of MM-141, a dual antibody inhibitor of IGF-1R and ErbB3. Cancer Res. 2015;75(15 suppl): abstr CT237-CT. doi:10.1158/1538-7445.am2015-ct237.
Habben K, Delmar P, Brownstein CM, Koehler W, Kuenkele K, Splesis O, et al. Investigation of predictive biomarkers for R1507, an anti-IGF1R antibody, in patients with advanced non-small cell lung cancer with progression after first-line chemotherapy. J Clin Oncol. 2011;29(suppl):abstr 7584.
Pavlicek A, Lira ME, Lee NV, Ching KA, Ye J, Cao J, et al. Molecular predictors of sensitivity to the insulin-like growth factor 1 receptor inhibitor Figitumumab (CP-751,871). Mol Cancer Ther. 2013;12(12):2929–39. doi:10.1158/1535-7163.MCT-13-0442-T.
Zha J, O’Brien C, Savage H, Huw LY, Zhong F, Berry L, et al. Molecular predictors of response to a humanized anti-insulin-like growth factor-I receptor monoclonal antibody in breast and colorectal cancer. Mol Cancer Ther. 2009;8(8):2110–21. doi:10.1158/1535-7163.MCT-09-0381.
Zhong H, Fazenbaker C, Chen C, Breen S, Huang J, Yao X, et al. Overproduction of IGF-2 drives a subset of colorectal cancer cells, which specifically respond to an anti-IGF therapeutic antibody and combination therapies. Oncogene. 2017;36(6):797–806. doi:10.1038/onc.2016.248.
Zinn RL, Gardner EE, Marchionni L, Murphy SC, Dobromilskaya I, Hann CL, et al. ERK phosphorylation is predictive of resistance to IGF-1R inhibition in small cell lung cancer. Mol Cancer Ther. 2013;12(6):1131–9. doi:10.1158/1535-7163.MCT-12-0618.
Asmane I, Watkin E, Alberti L, Duc A, Marec-Berard P, Ray-Coquard I, et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: a predictive biomarker for IGF-1R monoclonal antibody (ab) therapy in sarcomas. Eur J Cancer. 2012;48(16):3027–35. doi:10.1016/j.ejca.2012.05.009.
Shen K, Cui D, Sun L, Lu Y, Han M, Liu J. Inhibition of IGF-IR increases chemosensitivity in human colorectal cancer cells through MRP-2 promoter suppression. J Cell Biochem. 2012;113(6):2086–97. doi:10.1002/jcb.24080.
Rochester MA, Riedemann J, Hellawell GO, Brewster SF, Macaulay VM. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents in both PTEN wild-type and mutant human prostate cancer. Cancer Gene Ther. 2005;12(1):90–100. doi:10.1038/sj.cgt.7700775.
Haluska P, Hou X, Huang F, Harrington S, Greer A, Macedo L, et al. Complete IGF signaling blockade by the dual-kinase inhibitor, BMS-754807, is sufficient to overcome tamoxifen and Letrozole resistance in vitro and in vivo. Cancer Res. 2009;69(24 suppl):402. doi:10.1158/0008-5472.sabcs-09-402.
Plymate SR, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, et al. An antibody targeting the type I insulin-like growth factor receptor enhances the castration-induced response in androgen-dependent prostate cancer. Clin Cancer Res. 2007;13(21):6429–39. doi:10.1158/1078-0432.CCR-07-0648.
von Manstein V, Yang CM, Richter D, Delis N, Vafaizadeh V, Groner B. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr Signal Transduct Ther. 2013;8(3):193–202. doi:10.2174/1574362409666140206221931.
Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118(7):2609–19. doi:10.1172/JCI34588.
Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73(2):834–43. doi:10.1158/0008-5472.CAN-12-2066.
Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther. 2012;11(12):2644–53. doi:10.1158/1535-7163.MCT-12-0447.
Ireland L, Santos A, Ahmed MS, Rainer C, Nielsen SR, Quaranta V, et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016;76(23):6851–6863. doi:10.1158/0008-5472.CAN-16-1201.
Zeng X, Sachdev D, Zhang H, Gaillard-Kelly M, Yee D. Sequencing of type I insulin-like growth factor receptor inhibition affects chemotherapy response in vitro and in vivo. Clin Cancer Res. 2009;15(8):2840–9. doi:10.1158/1078-0432.CCR-08-1401.
Ramcharan R, Aleksic T, Kamdoum WP, Gao S, Pfister SX, Tanner J, et al. IGF-1R inhibition induces schedule-dependent sensitization of human melanoma to temozolomide. Oncotarget. 2015;6(37):39877–90. doi:10.18632/oncotarget.5631.
Matsumoto F, Valdecanas DN, Mason KA, Milas L, Ang KK, Raju U. The impact of timing of EGFR and IGF-1R inhibition for sensitizing head and neck cancer to radiation. Anticancer Res. 2012;32(8):3029–35.
Osuka S, Sampetrean O, Shimizu T, Saga I, Onishi N, Sugihara E, et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2013;31(4):627–40. doi:10.1002/stem.1328.
Simone BA, Dan T, Palagani A, Jin L, Han SY, Wright C, et al. Caloric restriction coupled with radiation decreases metastatic burden in triple negative breast cancer. Cell Cycle. 2016;15(17):2265–74. doi:10.1080/15384101.2016.1160982.
Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002;62(1):200–7.
Overholser J, Ambegaokar KH, Eze SM, Sanabria-Figueroa E, Nahta R, Bekaii-Saab T, et al. Anti-tumor effects of peptide therapeutic and peptide vaccine antibody co-targeting HER-1 and HER-2 in esophageal cancer (EC) and HER-1 and IGF-1R in triple-negative breast cancer (TNBC). Vaccines (Basel). 2015;3(3):519–43. doi:10.3390/vaccines3030519.
Bielen A, Perryman L, Box GM, Valenti M, de Haven BA, Martins V, et al. Enhanced efficacy of IGF1R inhibition in pediatric glioblastoma by combinatorial targeting of PDGFRalpha/beta. Mol Cancer Ther. 2011;10(8):1407–18. doi:10.1158/1535-7163.MCT-11-0205.
Dayyani F, Parikh NU, Varkaris AS, Song JH, Moorthy S, Chatterji T, et al. Combined inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS One. 2012;7(12):e51189. doi:10.1371/journal.pone.0051189.
Min HY, Yun HJ, Lee JS, Lee HJ, Cho J, Jang HJ, et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol Cancer. 2015;14:113. doi:10.1186/s12943-015-0392-3.
Wan X, Yeung C, Heske C, Mendoza A, Helman LJ. IGF-1R inhibition activates a YES/SFK bypass resistance pathway: rational basis for co-targeting IGF-1R and yes/SFK kinase in rhabdomyosarcoma. Neoplasia. 2015;17(4):358–66. doi:10.1016/j.neo.2015.03.001.
Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121(11):4311–21. doi:10.1172/JCI57909.
Flanigan SA, Pitts TM, Newton TP, Kulikowski GN, Tan AC, McManus MC, et al. Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clin Cancer Res. 2013;19(22):6219–29. doi:10.1158/1078-0432.CCR-13-0145.
Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013;3(5):548–63. doi:10.1158/2159-8290.CD-12-0446.
Cardillo TM, Trisal P, Arrojo R, Goldenberg DM, Chang CH. Targeting both IGF-1R and mTOR synergistically inhibits growth of renal cell carcinoma in vitro. BMC Cancer. 2013;13:170. doi:10.1186/1471-2407-13-170.
Di Cosimo S, Seoane J, Guzman M, Rojo F, Jimenez J, Anido J et al. Combination of the mammalian target of rapamycin (mTOR) inhibitor everolimus (E) with the insulin like growth factor-1-receptor (IGF-1-R) inhibitor NVP-AEW-541: A mechanistic based anti-tumor strategy. J Clin Oncol. 2005;23(16 suppl):abstr 3112.
Di Cosimo S, Sathyanarayanan S, Bendell JC, Cervantes A, Stein MN, Brana I, et al. Combination of the mTOR inhibitor ridaforolimus and the anti-IGF1R monoclonal antibody dalotuzumab: preclinical characterization and phase I clinical trial. Clin Cancer Res. 2015;21(1):49–59. doi:10.1158/1078-0432.CCR-14-0940.
Herkert B, Kauffmann A, Molle S, Schnell C, Ferrat T, Voshol H, et al. Maximizing the efficacy of MAPK-targeted treatment in PTENLOF/BRAFMUT melanoma through PI3K and IGF1R inhibition. Cancer Res. 2016;76(2):390–402. doi:10.1158/0008-5472.CAN-14-3358.
Flashner-Abramson E, Klein S, Mullin G, Shoshan E, Song R, Shir A, et al. Targeting melanoma with NT157 by blocking Stat3 and IGF1R signaling. Oncogene. 2016;35(20):2675–80. doi:10.1038/onc.2015.229.
Sanchez-Lopez E, Flashner-Abramson E, Shalapour S, Zhong Z, Taniguchi K, Levitzki A, et al. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene. 2016;35(20):2634–44. doi:10.1038/onc.2015.326.
Heilmann AM, Perera RM, Ecker V, Nicolay BN, Bardeesy N, Benes CH, et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res. 2014;74(14):3947–58. doi:10.1158/0008-5472.CAN-13-2923.
Miller ML, Molinelli EJ, Nair JS, Sheikh T, Samy R, Jing X, et al. Drug synergy screen and network modeling in dedifferentiated liposarcoma identifies CDK4 and IGF1R as synergistic drug targets. Sci Signal. 2013;6(294):ra85. doi:10.1126/scisignal.2004014.
Amin O, Beauchamp MC, Nader PA, Laskov I, Iqbal S, Philip CA, et al. Suppression of homologous recombination by insulin-like growth factor-1 inhibition sensitizes cancer cells to PARP inhibitors. BMC Cancer. 2015;15:817. doi:10.1186/s12885-015-1803-y.
Cao H, Dong W, Qu X, Shen H, Xu J, Zhu L, et al. Metformin enhances the therapy effects of Anti-IGF-1R mAb Figitumumab to NSCLC. Sci Rep. 2016;6:31072. doi:10.1038/srep31072.
O’Flanagan CH, O’Shea S, Lyons A, Fogarty FM, McCabe N, Kennedy RD, et al. IGF-1R inhibition sensitizes breast cancer cells to ATM-related kinase (ATR) inhibitor and cisplatin. Oncotarget. 2016;7(35):56826–56841. doi:10.18632/oncotarget.10862.
Mancarella C, Casanova-Salas I, Calatrava A, Ventura S, Garofalo C, Rubio-Briones J, et al. ERG deregulation induces IGF-1R expression in prostate cancer cells and affects sensitivity to anti-IGF-1R agents. Oncotarget. 2015;6(18):16611–22. doi:10.18632/oncotarget.3425.
Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352(6288) doi:10.1126/science.aad3018.
Maris C, D’Haene N, Trepant AL, Le MM, Sauvage S, Allard J, et al. IGF-IR: a new prognostic biomarker for human glioblastoma. Br J Cancer. 2015;113(5):729–37. doi:10.1038/bjc.2015.242.
Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, et al. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2014;32(19):2059–66. doi:10.1200/JCO.2013.54.4932.
Sclafani F, Kim TY, Cunningham D, Kim TW, Tabernero J, Schmoll HJ, et al. A randomized phase II/III study of Dalotuzumab in combination with Cetuximab and irinotecan in Chemorefractory, KRAS wild-type, metastatic colorectal cancer. J Natl Cancer Inst. 2015;107(12):djv258. doi:10.1093/jnci/djv258.
Ko AH, Murray J, Horgan KE, Dauer J, Curley M, Baum J et al. A multicenter phase II study of istiratumab (MM-141) plus nab-paclitaxel (A) and gemcitabine (G) in metastatic pancreatic cancer (MPC). J Clin Oncol. 2016;34(4S suppl): abstr TPS481.
Yamaoka T, Ohmori T, Ohba M, Arata S, Kishino Y, Murata Y, et al. Acquired resistance mechanisms to combination met-TKI/EGFR-TKI exposure in met-amplified EGFR-TKI resistant lung adenocarcinoma harboring an activating EGFR mutation. Mol Cancer Ther. 2016;15(12):3040–54. doi:10.1158/1535-7163.MCT-16-0313.
Davaadelger B, Duan L, Perez RE, Gitelis S, Maki CG. Crosstalk between the IGF-1R/AKT/mTORC1 pathway and the tumor suppressors p53 and p27 determines cisplatin sensitivity and limits the effectiveness of an IGF-1R pathway inhibitor. Oncotarget. 2016;7(19):27511–26. doi:10.18632/oncotarget.8484.
Park K, Cho KH, Lee KH, Su W, Kim S, Lin C et al. Phase Ib trial of afatinib and xentuzumab (BI 836845) in advanced NSCLC: dose-escalation and safety results. J Thorac Oncol. 2017;12(1, suppl):S1187–S1188.
Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell. 2015;27(1):109–22. doi:10.1016/j.ccell.2014.11.008.
Becker MA, Hou X, Tienchaianada P, Haines BB, Harrington SC, Weroha SJ, et al. Ridaforolimus (MK-8669) synergizes with Dalotuzumab (MK-0646) in hormone-sensitive breast cancer. BMC Cancer. 2016;16(1):814. doi:10.1186/s12885-016-2847-3.
Lamhamedi-Cherradi SE, Menegaz BA, Ramamoorthy V, Vishwamitra D, Wang Y, Maywald RL et al. IGF-1R and mTOR blockade: Novel resistance mechanisms and synergistic drug combinations for Ewing Sarcoma. J Natl Cancer Inst. 2016;108(12). doi:10.1093/jnci/djw182.
LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34(31):3803–15. doi:10.1200/JCO.2014.59.0018.
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–25. doi:10.1158/1078-0432.CCR-11-2381.
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370(6490):527–32. doi:10.1038/370527a0.
Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 1993;364(6435):308–13. doi:10.1038/364308a0.
Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–8. doi:10.1038/nature08768.
Sciacca L, Le MR, Vigneri R. Insulin analogs and cancer. Front Endocrinol (Lausanne). 2012;3:21. doi:10.3389/fendo.2012.00021.
Chan JY, LaPara K, Yee D. Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells. Oncogene. 2016;35(32):4235–43. doi:10.1038/onc.2015.488.
Brana I, Berger R, Golan T, Haluska P, Edenfield J, Fiorica J, et al. A parallel-arm phase I trial of the humanised anti-IGF-1R antibody dalotuzumab in combination with the AKT inhibitor MK-2206, the mTOR inhibitor ridaforolimus, or the NOTCH inhibitor MK-0752, in patients with advanced solid tumours. Br J Cancer. 2014;111(10):1932–44. doi:10.1038/bjc.2014.497.
Calvo E, Ma W, Tolcher AW, Hidalgo M, Soria J, Bahleda R et al. Phase (P) I study of PF-00299804 (PF) combined with figitumumab (FI; CP-751871) in patients (pts) with advanced solid tumors (ASTs). J Clin Oncol. 2010;28(15 suppl): abstr 3026.
Naing A, Kurzrock R, Burger A, Gupta S, Lei X, Busaidy N, et al. Phase I trial of cixutumumab combined with temsirolimus in patients with advanced cancer. Clin Cancer Res. 2011;17(18):6052–60. doi:10.1158/1078-0432.CCR-10-2979.
Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17(4):871–9. doi:10.1158/1078-0432.CCR-10-2621.
Macaulay VM, Middleton MR, Eckhardt SG, Rudin CM, Juergens RA, Gedrich R, et al. Phase I dose-escalation study of Linsitinib (OSI-906) and Erlotinib in patients with advanced solid tumors. Clin Cancer Res. 2016;22(12):2897-907. doi:10.1158/1078-0432.CCR-15-2218.
Leighl NB, Rizvi NA, de Lima LG Jr, Arpornwirat W, Rudin CM, Chiappori AA, et al. Phase 2 study of Erlotinib in combination with Linsitinib (OSI-906) or placebo in chemotherapy-naive patients with non-small-cell lung cancer and activating epidermal growth factor receptor mutations. Clin Lung Cancer. 2017;18(1):34–42.e2. doi:10.1016/j.cllc.2016.07.007.
Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, Tebbutt NC, Mitchell E, et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res. 2014;20(16):4240–50. doi:10.1158/1078-0432.CCR-13-2752.
Bendell JC, Jones SF, Hart L, Spigel DR, Lane CM, Earwood C, et al. A phase Ib study of linsitinib (OSI-906), a dual inhibitor of IGF-1R and IR tyrosine kinase, in combination with everolimus as treatment for patients with refractory metastatic colorectal cancer. Investig New Drugs. 2015;33(1):187–93. doi:10.1007/s10637-014-0177-3.
Wagner LM, Fouladi M, Ahmed A, Krailo MD, Weigel B, DuBois SG, et al. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology group. Pediatr Blood Cancer. 2015;62(3):440–4. doi:10.1002/pbc.25334.
Dasari A, Phan A, Gupta S, Rashid A, Yeung SC, Hess K, et al. Phase I study of the anti-IGF1R antibody cixutumumab with everolimus and octreotide in advanced well-differentiated neuroendocrine tumors. Endocr Relat Cancer. 2015;22(3):431–41. doi:10.1530/ERC-15-0002.
Baselga J, Morales S, Awada A, Blum J, Tan A, Ewertz M, et al. A phase 2 study of ridaforolimus (RIDA) and dalotuzumab (DALO) in estrogen receptor positive (ER+) breast cancer. Cancer Res. 2013;73(24 suppl)abstr: P2-16-04. doi:10.1158/0008-5472.sabcs13-p2-16-04.
Dean JP, Sprenger CC, Wan J, Haugk K, Ellis WJ, Lin DW, et al. Response of the insulin-like growth factor (IGF) system to IGF-IR inhibition and androgen deprivation in a neoadjuvant prostate cancer trial: effects of obesity and androgen deprivation. J Clin Endocrinol Metab. 2013;98(5):E820–8. doi:10.1210/jc.2012-3856.
Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res. 2005;65(5):1849–57. doi:10.1158/0008-5472.CAN-04-1837.
Robertson JF, Ferrero JM, Bourgeois H, Kennecke H, de Boer RH, Jacot W, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013;14(3):228–35. doi:10.1016/S1470-2045(13)70026-3.
Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–30. doi:10.1172/JCI41824.
Sharon SM, Pozniak Y, Geiger T, Werner H. TMPRSS2-ERG fusion protein regulates insulin-like growth factor-1 receptor (IGF1R) gene expression in prostate cancer: involvement of transcription factor Sp1. Oncotarget. 2016;7(32):51375–92. doi:10.18632/oncotarget.9837.
Wilson S, Chia SK. IGF-1R inhibition: right direction, wrong pathway? Lancet Oncol. 2013;14(3):182–3. doi:10.1016/S1470-2045(13)70019-6.
Cortes J, Martinez Janez N, Sablin MP, Perez-Fidalgo JA, Neven P, Hedayati E et al. Phase 1b/2 trial of BI 836845, an insulin-like growth factor (IGF) ligand-neutralizing antibody, combined with exemestane (Ex) and everolimus (Ev) in hormone receptor-positive (HR+) locally advanced or metastatic breast cancer (BC): primary phase 1b results. J Clin Oncol. 2016;34(suppl):abstr 530.
Basch E, Loblaw DA, Oliver TK, Carducci M, Chen RC, Frame JN, et al. Systemic therapy in men with metastatic castration-resistant prostate cancer: American Society of Clinical Oncology and Cancer Care Ontario clinical practice guideline. J Clin Oncol. 2014;32(30):3436–48. doi:10.1200/JCO.2013.54.8404.
Parker C, Gillessen S, Heidenreich A, Horwich A. Cancer of the prostate: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(suppl 5):v69–77. doi:10.1093/annonc/mdv222.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced Nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39. doi:10.1056/NEJMoa1507643.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28. doi:10.1056/NEJMoa1501824.
Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–50. doi:10.1016/S0140-6736(15)01281-7.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:10.1056/NEJMoa1003466.
Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(13):1270–1. doi:10.1056/NEJMc1509660.
Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908–18. doi:10.1016/S1470-2045(15)00083-2.
Wargo JA, Cooper ZA, Flaherty KT. Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer Discov. 2014;4(12):1377–86. doi:10.1158/2159-8290.CD-14-0477.
Andrews DW, Resnicoff M, Flanders AE, Kenyon L, Curtis M, Merli G, et al. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J Clin Oncol. 2001;19(8):2189–200. doi:10.1200/jco.2001.19.8.2189.
Resnicoff M, Sell C, Rubini M, Coppola D, Ambrose D, Baserga R, et al. Rat glioblastoma cells expressing an antisense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res. 1994;54(8):2218–22.
Trojan J, Johnson TR, Rudin SD, Ilan J, Tykocinski ML, Ilan J. Treatment and prevention of rat glioblastoma by immunogenic C6 cells expressing antisense insulin-like growth factor I RNA. Science. 1993;259(5091):94–7.
Anguela XM, Tafuro S, Roca C, Callejas D, Agudo J, Obach M, et al. Nonviral-mediated hepatic expression of IGF-I increases Treg levels and suppresses autoimmune diabetes in mice. Diabetes. 2013;62(2):551–60. doi:10.2337/db11-1776.
Bilbao D, Luciani L, Johannesson B, Piszczek A, Rosenthal N. Insulin-like growth factor-1 stimulates regulatory T cells and suppresses autoimmune disease. EMBO Mol Med. 2014;6(11):1423–35. doi:10.15252/emmm.201303376.
Johannesson B, Sattler S, Semenova E, Pastore S, Kennedy-Lydon TM, Sampson RD, et al. Insulin-like growth factor-1 induces regulatory T cell-mediated suppression of allergic contact dermatitis in mice. Dis Model Mech. 2014;7(8):977–85. doi:10.1242/dmm.015362.
Kooijman R, Coppens A. Insulin-like growth factor-I stimulates IL-10 production in human T cells. J Leukoc Biol. 2004;76(4):862–7.
Lu MC, Yu CL, Chen HC, Yu HC, Huang HB, Lai NS. Increased miR-223 expression in T cells from patients with rheumatoid arthritis leads to decreased insulin-like growth factor-1-mediated interleukin-10 production. Clin Exp Immunol. 2014;177(3):641–51. doi:10.1111/cei.12374.
Barrett JP, Minogue AM, Falvey A, Lynch MA. Involvement of IGF-1 and Akt in M1/M2 activation state in bone marrow-derived macrophages. Exp Cell Res. 2015;335(2):258–68. doi:10.1016/j.yexcr.2015.05.015.
Sprinzl MF, Puschnik A, Schlitter AM, Schad A, Ackermann K, Esposito I, et al. Sorafenib inhibits macrophage-induced growth of hepatoma cells by interference with insulin-like growth factor-1 secretion. J Hepatol. 2015;62(4):863–70. doi:10.1016/j.jhep.2014.11.011.
Somasundaram R, Zhang G, Wagner SN, Fukunaga-Kalabis M, Herlyn M. The role of tumor microenvironment in therapy resistance and melanoma progression. Cancer Res. 2015;75 (15 suppl):abstr 420.
USA Department of Health and Human Services Food and Drug Administration. Guidance for Industry: Adaptive Design Clinical Trials for Drugs and Biologics. 2010. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm201790.pdf
Acknowledgements
Research in the laboratory of VMM is supported by National Institute for Health Research (NIHR) Biomedical Research Centre, Cancer Research UK Oxford Cancer Research Centre, Breast Cancer Now, The Rosetrees Trust and UCARE-Oxford.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Fiona Scott, of GeoMed, an Ashfield company, part of UDG Healthcare plc, during the preparation of this article.
Conflict of Interest
Aaron Simpson and Wilfride Petnga have no conflicts of interest to declare. Valentine Macaulay has been a consultancy board member for Boehringer Ingelheim. Ulrike Weyer-Czernilofsky and Thomas Bogenrieder are employees of Boehringer Ingelheim RCV. Thomas Bogenrieder has stock ownership in Roche, Seattle Genetics, and Immunogen.
Additional information
Aaron Simpson and Wilfride Petnga are joint first authors
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/5D48F06008D136AC.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Simpson, A., Petnga, W., Macaulay, V.M. et al. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Targ Oncol 12, 571–597 (2017). https://doi.org/10.1007/s11523-017-0514-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-017-0514-5