
Abstract
Theoretical investigations have been carried out on the mechanism and kinetics for the reaction of CF3CHO + Cl using duallevel direct dynamics method. The potential energy surface information was obtained at the MC-QCISD/3//MP2/cc-pVDZ level and the kinetic calculations were done using variational transition state theory with interpolated single-point energy (VTST-ISPE) approach. The calculated results show that the reaction proceeds primarily via the H-abstraction channel, while the Cl-addition channel is unfavorable due to the higher barriers. The improved canonical variational transition-state theory (ICVT) with the small-curvature tunneling correction (SCT) was used to calculate the rate constants. The theoretical rate constants at room temperature are in general agreement with the experimental values. A three-parameter rate constant expression was fitted over a wide temperature range of 200–2000 K.
Similar content being viewed by others

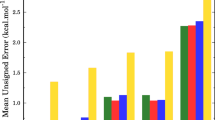
References
Wallington TJ, Schneider WF, Nielsen OJ, Sehested J, Worsnop DR, Bruyn WJ De, Shorter JA. Atmospheric chemistry and environmental impact of hydrofluorocarbons and hydrochlorofluorocarbons. In Miziolek AW, Tsang W, eds. Halon Replacements: Technology and Science. Washington DC: American Chemical Society, 1995. 16–30
Nelson L, Shanahan I, Sidebottom HW, Treacy J, Nielsen OJ. Kinetics and mechanism for the oxidation of 1, 1, 1-trichloroethane. Int J Chem Kinet, 1990, 22: 577–590
Mogelberg TE, Bilde M, Sehested J. Atmospheric chemistry of 1, 1, 1, 2-tetrachloroethane (CCl3CH2Cl): Spectrokinetic investigation of the CCl3CClHO2 radical, its reactions with NO and NO2, and atmospheric fate of the CCl3CClHO radical. J Phys Chem, 1996, 100: 18399–18407
Scollard DJ, Treacy JJ, Sidebottom HW, Balestra-Garcia C, Laverdet G, Le Bras G, Mac Leod H, Teton S. Rate constants for the reactions of hydroxyl radicals and chlorine atoms with halogenated aldehydes. J Phys Chem, 1993, 97: 4683–4688
Wallington TJ, Hurley MD. A kinetic study of the reaction of chlorine and fluorine atoms with CF3CHO at 295 ± 2 K. Int J Chem Kinet, 1993, 25: 819–824
Hurley MD, Wallington TJ, Sulbaek Andersen MP, Ellis DA, Martin JW, Mabury SA. Atmospheric chemistry of fluorinated alcohols: Reaction with Cl Atoms, OH radicals and atmospheric lifetimes. J Phys Chem A, 2004, 108: 1973–1979
Sulbaek Andersen MP, Nielsen OJ, Hurley MD, Ball JC, Wallington TJ, Stevens JE, Martin JW, Ellis DA, Mabury SA. Atmospheric chemistry of n-CxF2x+1CHO (x = 1, 3, 4): Reaction with Cl atoms, OH radicals and IR spectra of CxF2x+1C(O)O2NO2. J Phys Chem A, 2004, 108: 5189–5196
Dobe S, Khachatryan LA, Berces T. Kinetics of reactions of hydroxyl radicals with a series of aliphatic aldehydes. Ber Bunsen-Ges Phys Chem, 1989, 93: 847–852
Dibble TS, Francisco JS. Ab initio study of the reaction CF3CHO + X → CF3CO + HX (X = F, Cl). Chem Phys Lett, 1993, 215: 409–415
Truhlar DG. The Reaction Path, in the Reaction Path in Chemistry: Current Approaches and Perspectives. Dordrecht: Kluwer, 1995, 229
Truhlar DG, Garrett BC, Klippenstein SJ. Current status of transition-state theory. J Phys Chem, 1996, 100: 12771–12800
Hu WP, Truhlar DG. Factors affecting competitive ion-molecule reactions: ClO− + C2H5Cl and C2D5Cl via E2 and SN2 channels. J Am Chem Soc, 1996, 118: 860–869
POLYRATE, version 9. 3.1. Minneapolis: University of Minnesota. 2005
Truhlar DG, Garrett BC. Variational transition-state theory. Acc Chem Res, 1980, 13: 440–448
Truhlar DG, Brown FB, Steckler R, Isaacson AD. The representation and use of potential energy surfaces in the wide vicinity of a reaction path for dynamics calculations on polyatomic reactions. In Baer M, Ed. The Theory of Chemical Reaction Dynamics. Bocaraton, FL: CRC Press, 1985. 65
Truhlar DG, Garrett BC. Variational transition-state theory. Annu Rev Phys Chem, 1984, 35: 159–189
Chuang YY, Corchado JC, Truhlar DG. Mapped interpolation scheme for single-point energy corrections in reaction rate calculations and a critical evaluation of dual-level reaction path dynamics methods. J Phys Chem A, 1999, 103: 1140–1149
Lu DH, Truong TN, Melissas VS, Lynch GC, Liu YP, Garrett BC, Steckler R, Isaacson AD, Rai SN, Hancock GC, Lauderdale JG, Joseph T, Truhlar DG. POLYRATE 4: A new version of a computer program for the calculation of chemical reaction rates for polyatomics. Comput Phys Commun, 1992, 71: 235–262
Liu YP, Lynch GC, Truong TN, Lu DH, Truhlar DG, Garrett BC. Molecular modeling of the kinetic isotope effect for the [1,5]-sigmatropic rearrangement of cis-1,3-pentadiene. J Am Chem Soc, 1993, 115: 2408–2415
Gaussian 03, Revision A. 1. Pittsburgh, PA: Guassian, Inc. 2003
Woon DE, Dunning TH Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys, 1993, 98: 1358–1371
Lynch BJ, Truhlar DG. Robust and affordable multicoefficient methods for thermochemistry and thermochemical kinetics: The MCCM/3 suite and SAC/3. J Phys Chem A, 2003, 107: 3898–3906
Curtiss LA, Redfern PR. Gaussian-3 theory using reduced Møller-Plesset order. J Chem Phys, 1999, 110: 4703–4709
Garrett BC, Truhlar DG, Grev RS, Magnuson AW. Improved treatment of threshold contributions in variational transition-state theory. J Phys Chem, 1980, 84: 1730–1748
Huber KP, Herzberg G. Molecular Spectra and Molecular Structure. Constants of Diatomic Molecules. New York: Van Nostrand Reinhold Company, 1979
Pacey PD. Changing conceptions of activation energy. J Chem Educ, 1981, 58: 612–614
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gao, H., Wang, Y., Wang, Q. et al. Mechanistic study and kinetic properties of the CF3CHO + Cl reaction. Sci. China Chem. 55, 2197–2201 (2012). https://doi.org/10.1007/s11426-012-4745-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11426-012-4745-0