
Abstract
Introduction
A decline in mitochondrial function represents a key factor of a large number of inborn errors of metabolism, which lead to an extremely heterogeneous group of disorders.
Objectives
To gain insight into the biochemical consequences of mitochondrial dysfunction, we performed a metabolic profiling study in human skin fibroblasts using galactose stress medium, which forces cells to rely on mitochondrial metabolism.
Methods
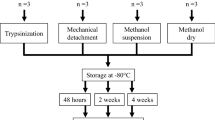
Fibroblasts from controls, complex I and pyruvate dehydrogenase (PDH) deficient patients were grown under glucose or galactose culture condition. We investigated extracellular flux using Seahorse XF24 cell analyzer and assessed metabolome fingerprints using NMR spectroscopy.
Results
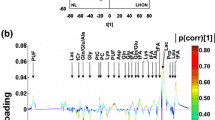
Incubation of fibroblasts in galactose leads to an increase in oxygen consumption and decrease in extracellular acidification rate, confirming adaptation to a more aerobic metabolism. NMR allowed rapid profiling of 41 intracellular metabolites and revealed clear separation of mitochondrial defects from controls under galactose using partial least squares discriminant analysis. We found changes in classical markers of mitochondrial metabolic dysfunction, as well as unexpected markers of amino acid and choline metabolism. PDH deficient cell lines showed distinct upregulation of glutaminolytic metabolism and accumulation of branched-chain amino acids, while complex I deficient cell lines were characterized by increased levels in choline metabolites under galactose.
Conclusion
Our results show the relevance of selective culture methods in discriminating normal from metabolic deficient cells. The study indicates that untargeted fingerprinting NMR profiles provide physiological insight on metabolic adaptations and can be used to distinguish cellular metabolic adaptations in PDH and complex I deficient fibroblasts.




Similar content being viewed by others

Change history
19 March 2019
The original version of this article contained an error in Table 2. The text in the second header line should read “GAL supernatant” and “GAL Medium” instead of “GLC supernatant” and “GLC Medium”. The corrected Table 2 is given below. The original article has been corrected.
References
Aguer, C., et al. (2011). Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE, 6, e28536. https://doi.org/10.1371/journal.pone.0028536.
Aguilar, J. A., Nilsson, M., Bodenhausen, G., & Morris, G. A. (2012). Spin echo NMR spectra without J modulation. Chemical Communications (Camb), 48, 811–813. https://doi.org/10.1039/c1cc16699a.
Baykal, A. T., Jain, M. R., & Li, H. (2008). Aberrant regulation of choline metabolism by mitochondrial electron transport system inhibition in neuroblastoma cells. Metabolomics, 4, 347–356. https://doi.org/10.1007/s11306-008-0125-3.
Beckonert, O., et al. (2010). High-resolution magic-angle-spinning NMR spectroscopy for metabolic profiling of intact tissues. Nature Protocols, 5, 1019–1032. https://doi.org/10.1038/nprot.2010.45.
Bluml, S., Seymour, K. J., & Ross, B. D. (1999). Developmental changes in choline- and ethanolamine-containing compounds measured with proton-decoupled (31)P MRS in in vivo human brain. Magnetic Resonance in Medicine, 42, 643–654.
Brown, G. K., Otero, L. J., LeGris, M., & Brown, R. M. (1994). Pyruvate dehydrogenase deficiency. Journal of Medical Genetics, 31, 875–879.
Dieterle, F., Ross, A., Schlotterbeck, G., & Senn, H. (2006). Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Analytical Chemistry, 78, 4281–4290. https://doi.org/10.1021/ac051632c.
DiMauro, S. (2004). Mitochondrial diseases. Biochimica et Biophysica Acta, 1658, 80–88. https://doi.org/10.1016/j.bbabio.2004.03.014.
Diserens, G., et al. (2017). Metabolic stability of cells for extended metabolomical measurements using NMR. A comparison between lysed and additionally heat inactivated cells. Analyst, 142, 465–471. https://doi.org/10.1039/c6an02195f.
Gropman, A. L. (2013). Neuroimaging in mitochondrial disorders. Neurotherapeutics, 10, 273–285. https://doi.org/10.1007/s13311-012-0161-6.
Harper, A. E., Miller, R. H., & Block, K. P. (1984). Branched-chain amino acid metabolism. Annual Review of Nutrition, 4, 409–454. https://doi.org/10.1146/annurev.nu.04.070184.002205.
Jansen, P. H., van der Knaap, M. S., & de Coo, I. F. (1996). Leber’s hereditary optic neuropathy with the 11 778 mtDNA mutation and white matter disease resembling multiple sclerosis: Clinical, MRI and MRS findings. Journal of the Neurological Sciences, 135, 176–180.
Johnson, M. T., Yang, H. S., & Patel, M. S. (2000). Targeting E3 component of alpha-keto acid dehydrogenase complexes. Methods in Enzymology, 324, 465–476.
Kirby, D. M., Crawford, M., Cleary, M. A., Dahl, H. H., Dennett, X., & Thorburn, D. R. (1999). Respiratory chain complex I deficiency: An underdiagnosed energy generation disorder. Neurology, 52, 1255–1264.
Koopman, W. J., Distelmaier, F., Smeitink, J. A., & Willems, P. H. (2013). OXPHOS mutations and neurodegeneration. EMBO Journal, 32, 9–29. https://doi.org/10.1038/emboj.2012.300.
Koopman, W. J., Willems, P. H., & Smeitink, J. A. (2012). Monogenic mitochondrial disorders. New England Journal of Medicine, 366, 1132–1141. https://doi.org/10.1056/NEJMra1012478.
Li, T., et al. (2017). Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metabolism, 25, 374–385. https://doi.org/10.1016/j.cmet.2016.11.005.
Li, Y., et al. (2016). PDHA1 gene knockout in prostate cancer cells results in metabolic reprogramming towards greater glutamine dependence. Oncotarget, 7, 53837–53852. https://doi.org/10.18632/oncotarget.10782.
Loeffen, J. L., et al. (2000). Isolated complex I deficiency in children: Clinical, biochemical and genetic aspects. Human Mutation, 15(200002), 123–134.
Lynch, C. J., & Adams, S. H. (2014). Branched-chain amino acids in metabolic signalling and insulin resistance. Nature Reviews Endocrinology, 10, 723–736. https://doi.org/10.1038/nrendo.2014.171.
Marroquin, L. D., Hynes, J., Dykens, J. A., Jamieson, J. D., & Will, Y. (2007). Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicological Sciences, 97, 539–547. https://doi.org/10.1093/toxsci/kfm052.
Mastorodemos, V., Zaganas, I., Spanaki, C., Bessa, M., & Plaitakis, A. (2005). Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. Journal of Neuroscience Research, 79, 65–73. https://doi.org/10.1002/jnr.20353.
Mitochondrial Medicine Society’s Committee on, D., et al. (2008). The in-depth evaluation of suspected mitochondrial disease. Molecular Genetics and Metabolism, 94, 16–37. https://doi.org/10.1016/j.ymgme.2007.11.018.
Moestue, S., Sitter, B., Bathen, T. F., Tessem, M. B., & Gribbestad, I. S. (2011). HR MAS MR spectroscopy in metabolic characterization of cancer. Current Topics in Medicinal Chemistry, 11, 2–26.
Morvan, D., & Demidem, A. (2018). NMR metabolomics of fibroblasts with inherited mitochondrial Complex I mutation reveals treatment-reversible lipid and amino acid metabolism alterations. Metabolomics, 14, 55. https://doi.org/10.1007/s11306-018-1345-9.
Nicholson, J. K., Lindon, J. C., & Holmes, E. (1999). ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica, 29, 1181–1189. https://doi.org/10.1080/004982599238047.
Robinson, B. H. (1998). Human complex I deficiency: Clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochimica et Biophysica Acta, 1364, 271–286.
Robinson, B. H., MacKay, N., Chun, K., & Ling, M. (1996). Disorders of pyruvate carboxylase and the pyruvate dehydrogenase complex. Journal of Inherited Metabolic Diseases, 19, 452–462.
Robinson, B. H., Petrova-Benedict, R., Buncic, J. R., & Wallace, D. C. (1992). Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochemical Medicine and Metabolic Biology, 48, 122–126.
Rossignol, R., Gilkerson, R., Aggeler, R., Yamagata, K., Remington, S. J., & Capaldi, R. A. (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Research, 64, 985–993.
Smeitink, J. A. (2003). Mitochondrial disorders: Clinical presentation and diagnostic dilemmas. Journal of Inherited Metabolic Diseases, 26, 199–207.
Smeitink, J. A., Zeviani, M., Turnbull, D. M., & Jacobs, H. T. (2006). Mitochondrial medicine: A metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metabolism, 3, 9–13. https://doi.org/10.1016/j.cmet.2005.12.001.
Udhane, S. S., et al. (2017). Combined transcriptome and metabolome analyses of metformin effects reveal novel links between metabolic networks in steroidogenic systems. Scientific Reports, 7, 8652. https://doi.org/10.1038/s41598-017-09189-y.
Vafai, S. B., & Mootha, V. K. (2013). Medicine. A common pathway for a rare disease? Science, 342, 1453–1454. https://doi.org/10.1126/science.1248449.
Vermathen, M., Diserens, G., Vermathen, P., & Furrer, J. (2017). Metabolic profiling of cells in response to drug treatment using (1)H high-resolution magic angle spinning (HR-MAS) NMR spectroscopy. Chimia, 71, 124–129. https://doi.org/10.2533/chimia.2017.124.
Vermathen, M., Paul, L. E., Diserens, G., Vermathen, P., & Furrer, J. (2015). 1H HR-MAS NMR based metabolic profiling of cells in response to treatment with a hexacationic ruthenium metallaprism as potential anticancer drug. PLoS ONE, 10, e0128478. https://doi.org/10.1371/journal.pone.0128478.
Wishart, D. S., et al. (2013). HMDB 3.0—the human metabolome database in 2013. Nucleic Acids Research, 41, D801–D807. https://doi.org/10.1093/nar/gks1065.
Yang, C., et al. (2014). Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Molecular Cell, 56, 414–424. https://doi.org/10.1016/j.molcel.2014.09.025.
Zhou, X., & Thompson, J. R. (1996). Regulation of glutamate dehydrogenase by branched-chain amino acids in skeletal muscle from rats and chicks. The International Journal of Biochemistry and Cell Biology, 28, 787–793.
Acknowledgements
We thank André Schaller (Division of Human Genetics and Department of Paediatrics, Inselspital, Bern) for providing the genetic characteristics of the patient fibroblasts.
Funding
This work was supported by a grant from the Batzebär foundation of the children’s university hospitals Bern to JMN.
Author information
Authors and Affiliations
Contributions
DH, AF, GD, SK, PV, and JMN conceived the work and designed the experiments; DH, AF, and SK cultured cells, performed metabolic flux experiments and analyzed the data. DH and GD performed NMR analysis of cells or supernatant and analyzed the data. PV and JMN provided experimental advice and overall guidance. DH, AF, GD, SK, PV and JMN wrote the manuscript or revised it critically for important intellectual content. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the 1964 Helsinki declaration and its later amendments and approved by the Ethics Committee of the University Hospital of Bern.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: The errors in the Table 2 have been corrected.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hertig, D., Felser, A., Diserens, G. et al. Selective galactose culture condition reveals distinct metabolic signatures in pyruvate dehydrogenase and complex I deficient human skin fibroblasts. Metabolomics 15, 32 (2019). https://doi.org/10.1007/s11306-019-1497-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-019-1497-2