
Abstract
Plasmodium falciparum is the causative agent of the most dangerous form of malaria in humans. It has been reported that the P. falciparum genome encodes for a single ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase), an enzyme that hydrolyzes extracellular tri- and di-phosphate nucleotides. The E-NTPDases are known for participating in invasion and as a virulence factor in many pathogenic protozoa. Despite its presence in the parasite genome, currently, no information exists about the activity of this predicted protein. Here, we show for the first time that P. falciparum E-NTPDase is relevant for parasite lifecycle as inhibition of this enzyme impairs the development of P. falciparum within red blood cells (RBCs). ATPase activity could be detected in rings, trophozoites, and schizonts, as well as qRT-PCR, confirming that E-NTPDase is expressed throughout the intraerythrocytic cycle. In addition, transfection of a construct which expresses approximately the first 500 bp of an E-NTPDase-GFP chimera shows that E-NTPDase co-localizes with the endoplasmic reticulum (ER) in the early stages and with the digestive vacuole (DV) in the late stages of P. falciparum intraerythrocytic cycle.
Similar content being viewed by others

Introduction
Malaria is one of the most lethal parasitic human diseases in the developing world, causing about half a million deaths annually [1]. Its etiological agent belongs to the genus Plasmodium, and among these, Plasmodium falciparum is the one responsible for the most severe form of the disease [2]. It is well established that the signs and classic symptoms of malaria are due to the intraerythrocytic stages of the Plasmodium lifecycle (Fig. 1) [3]. There are reported cases of parasite resistance to all available anti-malarial drugs, and the understanding of the parasite physiology and signaling events will help to identify new drugs targets [4–6].

Plasmodium falciparum intraerythrocytic cycle. The figure shows the asexual stages of P. falciparum inside the RBCs. After the merozoite invasion, the parasite matures in distinct developmental stages, passing from the ring, through the trophozoite, to the schizont form. The rupture of a schizont-infected RBC releases more merozoites that will infect a new erythrocyte, starting a new cycle of replication
Components of the signaling machinery are being considered potential drug targets in malaria parasites. It is well known that Plasmodium is able to convert external stimuli into intracellular responses, [7–10].
The E-NTPDases, also called apyrases, are responsible for degradation of extracellular tri- and di-phosphate nucleotides and participates in parasite purine salvage pathway and purinergic signaling [11]. Specifically in P. falciparum, purinergic signaling has already been shown to participate in parasite lifecycle [12–14]. We demonstrate that P. falciparum is able to respond to ATP with a rise in intracellular calcium concentration. Additionally, depletion of ATP from the media was able to block parasite invasion of RBCs, pointing to a participation of purinergic signaling in this process [13].
In a more recent study from our group, we showed that in the rodent malaria, parasites P. berghei and P. yoelii addition of extracellular ATP also led to an increase in cytosolic calcium and this rise was blocked by purinergic antagonists. Incubation of P. berghei with the purinergic blocker KN-62 was able to change the MSP-1 processing profile and the pattern of parasite distribution in the erythrocytic cycle [15].
Previous work demonstrates that E-NTPDase activity is related with infectivity, virulence, and purine acquisition in many pathogenic protozoan parasites [16–22]. Santos et al. (2009) showed that the ecto-NTPDase inhibitors suramin, ARL67156, and gadolinium were capable of impair the in vitro infectivity of T. cruzi trypomastigotes. In another study, Bisaggio et al. (2003) observed that the ecto-ATPase activity of T. cruzi is about 20 times greater in trypomastigotes, as compared with epimastigotes. Additionally, the ecto-ATPase over-expression was followed by an increase in the adhesion of epimastigotes to resident macrophages [22].
In the apicomplexan relative T. gondii, two isozymes were found capable of hydrolyzing extracellular nucleotides (NTPase I and NTPase II). However, while the gene encoding NTPase II was found in all T. gondii species, the highly active enzyme NTPase I was only found in the virulent strain of T. gondii [18]. Activation of this enzyme by reducing agents leads to depletion of host cell ATP and parasite exit from host cells [23]. In Leishmania, it was shown that the more virulent parasite L. amazonensis hydrolyzes more ATP, ADP, and AMP than the other Leishmania species does [19]. The L. infantum NTPDase-2 functions as a genuine enzyme from the E-NTPDase/CD39 family being able to hydrolyze a wide variety of triphosphate and diphosphate nucleotides [21]. In the specie L. (V.) braziliensis, parasites with high ecto-nucleotidase activity are able to inhibit macrophage microbicidal activity, thus modulating the host immune response [24].
The genome database of P. falciparum predicted a gene encoding for a possible E-NTPDase (PF3D7_1431800) [25]. However, the activity of this enzyme has not been described [26]. In this work, we show that incubation of P. falciparum with known E-NTPDase inhibitors affects parasite development within RBCs, whereas ATPase activity points to a distinct capacity of ATP hydrolysis between the parasite stages. Quantification of apyrase mRNA by qRT-PCR shows that this enzyme is more expressed in trophozoites compared with rings and schizont stages. Co-localization studies performed using an N-terminal apyrase-GFP chimera clearly visualizes the fluorescence to the applied ER tracker suggesting a localization of the apyrase in the ER. This is the first report of apyrase activity in P. falciparum and consists in an important step towards the elucidation of E-NTPDase role in its asexual cycle.
Materials and methods
Reagents
All cell culture reagents were obtained from Cultilab (Brazil). Suramin, ARL 67156, and gadolinium chloride were purchased from Sigma Aldrich (St. Louis, MO).
Parasite cultivation and synchronization
P. falciparum, 3D7 strain, was maintained in continuous culture in adult human red blood cells [27], and the synchronization was achieved by sorbitol treatment [28].
Incubation of P. falciparum with E-NTPDases inhibitors
The parasitemia of a P. falciparum synchronous ring culture was adjusted to 1%, and the parasites were cultivated in a 48-well plate in the presence of 100 or 500 μM of the E-NTPDase inhibitors suramin, gadolinium chloride, and ARL 67156 for 48 h. An aliquot was collected from each well at different time points, and the parasitemia was assessed by flow cytometry as previously described [29]. The results were obtained from three independent experiments in triplicate.
Enzymatic assays
Isolated parasites from a synchronized culture of rings, trophozoites, or schizonts were obtained by adding saponin (SIGMA) to a final concentration of 0.05%. Following centrifugation at 8000 rpm at 4 °C for 8 min, erythrocyte ghosts were removed and the parasite pellets were washed twice using buffer M (in mM 116 NaCl, 5.4 KCl, 0.8 MgSO4, 5.5 D-glucose, 50 MOPS, 2 CaCl2) for 2 min at 10,000 rpm to remove any insoluble material. For the experiments using erythrocyte membranes, 50 μL of RBC pellet was resuspended in 500 μL of hyposmotic buffer for 10 min at RT. Following centrifugation at 6000 rpm at 4 °C for 6 min, membrane pellet was washed twice using the same buffer and kept on ice until the beginning of the experiment.
Apyrase activity was measured for 1 h at 37 °C, in the presence of 1 mM ATP and E-NTPDase inhibitors in a final volume of 80 μL of buffer M. The reaction started with the addition of 107 parasites or erythrocyte membranes. Parasite and erythrocyte membrane proteins were quantified by the Bradford method assay [30]. The amount of inorganic phosphate (Pi) released was measured as described by Ekman et al. 1993 [31].
Quantification of Pfapyrase expression by qRT-PCR
The total RNA was extracted from a synchronized culture of rings, trophozoites, and schizonts using TRIzol®. The cDNA synthesis was performed using 500 ng of total RNA and the Superscript II kit (Invitrogen) as described in the manufacturer’s protocol. Quantification of apyrase expression was performed by SYBR Green using a quantitative real-time PCR (qRT-PCR) in a 7300 Real-Time PCR system (Applied Biosystems). The sequences of used primers are provided in Table 1 (Pfapyrase and Seryl-tRNA synthetase control). The relative change in the amount of apyrase mRNA was determined by the 2Δct formula. The seryl-tRNA synthetase gene was amplified and used as normalizer. The experiments were performed in triplicate through three independent experiments.
Cloning and transfection of the GFP-fusion construct
The open reading frame (ORF) encoding Pfapyrase (PF3D7_1431800) was amplified by reverse transcriptase polymerase chain reaction (RT-PCR) (SuperScript III One-Step RT-PCR System, Invitrogen) using P. falciparum 3D7 total RNA. The sequences of the used primers are provided in Table 1 (Pfapyrase-GFP). The obtained PCR product (519 bp) was cloned in front of gfp via KpnI and AvrII restriction sites into the transfection vector pARL 1a- [32]. The nucleotide sequence was confirmed by automated sequencing before transfecting the plasmids into P. falciparum. Transfection was performed into ring stage-infected RBC. The selection of transgenic parasites was done by adding 5 nM of the selection drug WR99210.
Microscopy analysis of GFP-fusion construct
Live parasites were analyzed by fluorescent microscopy using an Axio Imager M2 microscope (Zeiss) equipped with an AxioCam HRC digital camera (Zeiss). Parasites were incubated with 10 μg/mL HOECHST 33342 (Invitrogen) to visualize the nucleus and 2 mM of ER-Tracker™ Red BODIPY-TR (Invitrogen) to show co-localization with the ER. The images were analyzed with the AxioVision 4.8 software.
Statistical analyses
Analyses were performed by t test or one-way analysis of variance (ANOVA) test followed by post hoc analysis by the Dunnett’s comparison test using GraphPad Prism software.
Results
E-NTPDases from different parasites have already been shown to participate in the invasion process of host cells [16, 19, 33, 34]. In order to assess whether the P. falciparum E-NTPDase is important for parasite development, we incubated P. falciparum with the E-NTPDase inhibitors suramin, ARL 67156, and gadolinium for 48 h and measured the parasitemia at different time points by flow cytometry (Fig. 2).

Effect of E-NTPDase inhibitors in intraerythrocytic development and invasion of RBCs by Plasmodium falciparum. Parasites in ring stage were incubated with the inhibitors and the parasitemia was measured after a 6, b 20, c 30, and d 48 h. A representative dot plot is presented at the bottom of each figure. Normalized data were analyzed statistically by ANOVA with Dunnett’s post test, bar graph means, and S.D. of three independent experiments. *p < 0.05; **p < 0.01
Suramin was able to negatively affect parasite development after 6, 20, 34, and 48 h of incubation at both applied concentrations (Fig. 2a–d). Suramin, a naftilurea polysulfone compound, has already been shown to partially inhibit the Ecto-ATPase and NTPDase-1 of T. cruzi [16, 22], as the ecto-apyrase of Torpedo electric organ [35]. Specifically, this inhibitor impaired erythrocyte invasion by P. falciparum; however, this effect was related to inhibition of MSP-1 processing and purinergic receptors located on parasite cell surface [12, 13].
Gadolinium was also capable of inhibiting the E-NTPDase from Torpedo electric organ and ecto-ATPase of T. cruzi [16, 36]. In our study, this compound inhibited P. falciparum development within erythrocytes after 20, 34, and 48 h of incubation at 500 μM (Fig. 2b–d).
ARL 67156 (6-N, N-diethyl-bc-dibromomethylene-d-adenosine-5-triphosphate), originally named FPL 67156, is described as a selective inhibitor of ecto-ATPase activity from blood cells and is able to inhibit the NTPDase-1 of T. cruzi [16, 37]. Similar to gadolinium, ARL67156 affects parasite development after 20 and 34 h of incubation at 500 μM (Fig. 2b, c). P. falciparum development within erythrocytes was impaired after 48 h at both tested concentrations (Fig. 2d).
In order to characterize the external ATPase activity, isolated parasites at different asexual stages (Fig. 3a) were incubated with 1 mM ATP in the presence of known E-NTPDase inhibitors (Fig. 3b). At ring stage, the parasites were able to release approximately 285.65 μM (±73.17, n = 4) of inorganic phosphate (Pi). Suramin and gadolinium were responsible for 52.01 μM (±29.72, n = 4) (81.79% inhibition) and 87.39 μM (±32.35, n = 4) (69.40% inhibition) of inorganic phosphate release, respectively. ARL 67156 had no inhibitory effect, releasing approximately 235.51 μM (±47.92, n = 4) of Pi.

ATPase activity in the P. falciparum intraerythrocytic cycle. a A micrograph showing the parasites (rings, trophozoites, and schizonts) utilized in the experiment. b 107 isolated parasites were incubated in the presence of 1 mM ATP and the E-NTPDase inhibitors suramin, gadolinium, and ARL 67156 for 1 h at 37 °C. The ATP degradation was measured by the amount of inorganic phosphate released. c The total amount of protein from 107 parasites was measured using the Bradford assay. d ATPase activity from erythrocyte membranes. RBC ghosts were obtained through incubation with hyposmotic buffer. Different concentrations of RBC membranes were incubated with 1 mM ATP for 1 h at 37 °C. The ATP degradation was measured by the amount of inorganic phosphate released. Data were analyzed statistically by ANOVA with the Dunnett’s post test, bar graph means, and S.D. of at least three independent experiments. *p < 0.01
In trophozoites, the ATPase activity was lower compared to the ring stage. After 1 h of incubation, hydrolysis of ATP released approximately 109.77 μM (±32.86, n = 5) of Pi. Both, suramin and gadolinium, block the ATP degradation (20.17 μM ± 23.08, n = 5) (81.62% inhibition) and (19.69 μM ± 12.43, n = 5) (82.06% inhibition), respectively, while no effect was observed in the presence of ARL 67156 (112.17 μM ± 30.57, n = 5). Similar results were obtained with schizonts (105.68 μM ± 36.55, n = 4). Suramin and gadolinium have a blocking effect ((41.33 μM ± 24.63, n = 4) (60.89% inhibition) and (39.39 μM ± 24.87, n = 4) (64.88% inhibition) respectively), whereas no inhibition was observed with ARL 67156 (120.21 μM ± 17.91, n = 4).
The total amount of proteins increase as the parasite develops (Fig. 3c). This fact is not surprising. P. falciparum changes its morphology during the asexual cycle, with a size increase from ring to trophozoite and then to schizont form. Interestingly, our results show that despite a higher quantity of proteins in trophozoites and schizonts, the E-NTPDase activity was higher in the ring stage, suggesting that the P. falciparum apyrase expression does not follow the same pattern.
It has already been shown that erythrocyte membranes have enzymes from the CD39 (ecto-apyrase) family [38]. Isolation of free parasites requires removal of RBC’s membrane. Despite several washes of free parasites to avoid contamination with RBC’s membrane, we decide to measure the ATPase activity of non-infected erythrocyte ghosts to ensure that the Pi release was only due to the E-NTPDase activity of the parasites. To assess this, we incubated RBCs in a hyposmotic buffer to remove its intracellular content and measured the amount of inorganic phosphate released by the erythrocyte membrane. Our results demonstrate that RBC ghosts are able to hydrolyze ATP (Fig. 3d). The phosphate release was detected at all tested concentrations from 5 to 200 μg of total protein; however, the ATP hydrolysis is much smaller than that presented by the parasites.
The expression profile of P. falciparum apyrase has already been shown in PlasmoDB [39]. Previous results demonstrate that this enzyme is expressed throughout the intraerythrocytic cycle, showing an increased expression in trophozoites [40]. However, these results were obtained by microarray analysis in which hundreds of genes are analyzed simultaneously.
In order to evaluate more specifically the expression profile of this enzyme, we decide to perform a qRT-PCR using specific primers for Pfapyrase. Our data show the apyrase is more expressed in trophozoites than in rings and schizonts, a pattern that is similar with previous results (Fig. 4). Interestingly, previous data (Fig. 3) show a higher ATPase activity in the ring stage, while the expression profile demonstrates that apyrase is more expressed in trophozoites. This fact could be justified by post-transcriptional regulation of gene expression. Specifically for the Pfapyrase gene in the ring stage, it was showed that its mRNA has a longer half-life (approximately 2-fold change) when compared with apyrase mRNA from trophozoites and schizonts [41]. As a consequence of mRNA stability, the levels of E-NTPDase in the ring stage could become higher, leading to a bigger amount of the enzyme in the parasite surface. These variations usually are related to the physiological role of some genes, determining the levels of gene expression.

Expression profile of P. falciparum apyrase throughout the intraerythrocytic cycle. Total RNA was extracted from a synchronous culture of wild-type 3D7 parasites (rings, trophozoites, and schizonts) and used to synthesize the complementary DNA (cDNA). A qRT-PCR was performed to measure the expression of apyrase mRNA. Data were analyzed statistically by t test, bar graph means, and S.D. of least three independent experiments. *p < 0.05
E-NTPDases are usually located in the cellular surface or secreted in the extracellular milieu [42]. Due the lack of information about this enzyme in P. falciparum, we decided to investigate its localization within the parasite. For this, we cloned the first 519 bp of the 5′terminus of the gene and tagged this sequence with the green fluorescent protein (GFP) (Fig. 5b). As can be seen, the P. falciparum apyrase is expressed throughout the asexual cycle (Fig. 5a). However, its localization changes as the parasite develops. In the ring and trophozoite stages, the P. falciparum apyrase co-localizes with the endoplasmic reticulum, also surrounding the parasite nucleus. As the parasite grows and forms several merozoites, a process known as schizogony (Fig. 1), the enzyme changes its localization being translocated to the digestive vacuole (Fig. 5a).

Localization of PfApyrase-GFP via live cell imaging. a The parasites were stained with HOECHST 33342 and ER-Tracker™ Red BODIPY-TR to visualize the nucleus and the ER, respectively. Different developmental stages of intraerythrocytic P. falciparum are shown to demonstrate the different localizations. The white arrows indicate the localization of the digestive vacuole within P. falciparum. b Cloning strategy to construct the E-NTPDase-GFP expressing P. falciparum. The first N-terminal 519 nucleotides of the apyrase gene were cloned in front of the green fluorescent protein (GFP). The plasmid was used to transfect the wild-type 3D7 P. falciparum strain, resulting in a transgenic line expressing the E-NTPDase tagged with GFP
Analysis of the P. falciparum E-NTPDase gene revealed the presence of two putative transmembrane domains located at the N- and C-terminus of the protein (data not show); however, no signal peptide has been predicted [39]. In the last step of the intraerythrocytic replication, the P. falciparum E-NTPDase changes its localization, being present in the digestive vacuole. A possible explanation relies in the fact that in some proteins, the N-terminal sequence works as a signal peptide, however without being cleaved [43, 44]. We propose that in the absence of the complete protein sequence, particularly the C-terminal transmembrane domain, our chimeric protein is retained in the Plasmodium ER, being translocated in the end of the replication cycle to the digestive vacuole.
Discussion
In order to assess the importance of E-NTPDase in P. falciparum lifecycle inside RBCs, we incubated infected erythrocytes with known apyrase inhibitors. Among all tested drugs, suramin, a naftilurea polysulfone compound, had the higher effect impairing parasite growth after 6, 20, 34, and 48 h. Despite its broad action, we believe that suramin is blocking the P. falciparum E-NTPDase, since data presented in Fig. 3b show an inhibition of ATP degradation in the presence of this drug. Gadolinium had a smaller effect in inhibiting the Plasmodium E-NTPDase. We could observe a decrease in the parasitemia after 20, 34, and 48 h only at the 500 μM concentration. These findings are in agreement with previous results in T. cruzi where gadolinium was able to inhibit the infectivity of this parasite in approximately 65% at 300 μM [16]. The selective inhibitor of ecto-ATPases ARL 67156 had a similar effect to gadolinium with a blocking effect in the parasitemia after 20 and 34 h at 500 μM. After 48 h of incubation, both concentrations of ARL 67156 were able to block the parasite development within RBCs. Interestingly, this drug was also able to impair the infectivity of T. cruzi in about 42% at 300 μM [16]. These findings show the effectiveness of the tested drugs, pointing to a participation of E-NTPDases in Plasmodium intraerythrocytic cycle.
E-NTPDase activity has already been characterized in several pathogenic protozoa, including Leishmania sp., T. cruzi, and T. gondii [16–19, 45–48]. The data presented in the literature suggest roles for E-NTPDases in parasite biology and disease pathogenesis [49]. Cell-surface located E-NTPDases play a key role in regulating purinergic signaling (ATP and other nucleotides acting as signaling molecules) [50]. Thus, by regulating purinergic signaling, the E-NTPDases of parasites are thought to influence a wide range of cellular functions as vascular homeostasis, nucleotide sugar transport, purine salvage, inflammation, and immune response [49].
Phylogenetic analysis revealed that P. falciparum E-NTPDase appears to be distinct from apyrases of other apicomplexan parasites, being similar to human and Schistosoma mansoni E-NTPDase [49]. This observation is particularly intriguing and suggests that this enzyme may play a different role in P. falciparum biology [26]. Comparative genomics of P. falciparum and P. vivax revealed a new subset of 15 genes that were considered novel candidates potentially linked to human severe malaria [51]. Included in this new subset, the E-NTPDase was also found to be exclusive of P. falciparum, being absent in other species of Plasmodium. This fact is extremely important and points toward an increased participation of this enzyme in the severe cases of malaria [51].
Despite that malaria is considered the most lethal parasitic disease, there is no data yet reporting nucleotidase activity of this protein in P. falciparum. Here, we show for the first time the presence of an ecto-enzyme in this parasite capable of hydrolyzing ATP at detectable rates. A higher ATPase activity in rings compared to trophozoites and schizonts was observed. Suramin and gadolinium blocked the ATP degradation at all parasite stages, while ARL 67156 had no effect in ATP hydrolysis. These data indicate that E-NTPDase could act providing purine precursors, since P. falciparum lack the ability to synthesize the purine ring de novo and rely on getting purines through the salvage pathway [52]. However, this does not rule out the participation of apyrase in other functions, for example as an adhesion molecule. The phylogenetic distance between P. falciparum and other aplicomplexan parasites E-NTPDases and the presence of apyrase only in P. falciparum among the Plasmodium species suggest that this enzyme could participate in virulence-associated events.
To rule out the possibility of contaminant ATPase activity arising from RBC membranes, we carried out the measurement of ATP degradation in erythrocyte ghosts. ATP hydrolysis was detected from 5 to 200 μg of total membrane proteins. However, even the highest ATP degradation at 200 μg total protein (approximately 12 μM) has a negligible value when compared to P. falciparum parasites.
Analyses of P. falciparum transcriptome showed that apyrase is more expressed in trophozoites [40]. To further confirm this, we performed a more specific assay to study the E-NTPDase expression profile in P. falciparum. By quantitative RT-PCR, we show that P. falciparum apyrase has a higher expression in trophozoites, followed by schizonts and rings (the last one having the smaller quantity of apyrase mRNA among the asexual stages). This result is in agreement with previous data from transcriptome analysis and points to a distinct pattern of E-NTPDase expression within the parasite lifecycle.
Interestingly, the ATPase activity profile, where rings have the highest ATP hydrolysis rate (Fig. 3b), does not match with the apyrase expression profile, in which trophozoites present a larger amount of apyrase (Fig. 4). Such discrepancy could be due to a post-transcriptional regulation of E-NTPDase gene expression. Previous results showed that the half-life apyrase mRNA from the ring stage is longer than the one from trophozoites and schizonts [41]. This higher mRNA stability could induce a bigger amount of enzyme in parasite surface, leading to a higher ATP degradation rate. However, more experiments are needed to prove this hypothesis.
E-NTPDases are known for its ecto-localization in the cell, although some enzymes could also be anchored in the membrane of organelles [42]. Bioinformatic analysis of P. falciparum E-NTPDase revealed the presence of two putative transmembrane domains located at the N- and C-terminus of the protein putting this enzyme in the group of membrane-bound E-NTPDases. To confirm this, we decided to clone the N-terminus of the gene in front of the sequence of the GFP protein. Transfection of P. falciparum with this construction showed that in the early stages (rings and trophozoites), the apyrase is in the parasite ER, as the enzyme co-localizes with the endoplasmic reticulum marker ER-Tracker (molecular probes). As the parasite develops and reaches the schizont stage, the E-NTPDase is gradually translocated to the digestive vacuole.
As mentioned before, E-NTPDases are mostly located outside the cell and the P. falciparum apyrase has two transmembrane domains that might support the same distribution in the parasite plasma membrane. Furthermore, the data presented here also shows the presence of an enzyme in the parasite surface able to hydrolyze ATP that is inhibited by known E-NTPDase inhibitors. This data highlight the importance of the complete protein sequence for the correct placement of the enzyme. The lack of C-terminal transmembrane sequence is impairing the localization of P. falciparum E-NTPDase. This behavior has already been reported in the literature, wherein an alternative C-terminal splicing pattern was able to provide distinctive catalytic properties, cellular distribution, and enzyme regulation of rat NTPDase2 [53]. Experiments performed with the entire protein tagged with GFP are needed to confirm this hypothesis and will clarify the correct localization of P. falciparum apyrase.
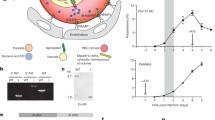
To the best of our knowledge, this is the first report of P. falciparum E-NTPDase and indicates that the E-NTPDase is important for parasite survival inside the host cell. ATPase activity could be detected in all parasite stages within the RBC, being higher in the ring stage, while mRNA profile reveals that trophozoites express more the enzyme than the rings and schizonts. The cellular distribution of an apyrase-GFP chimera shows that this enzyme is retained in the ER and is translocated to digestive vacuole at the end of parasite replication. As mentioned before, the presence of E-NTPDase only in P. falciparum among all Plasmodium species suggests that apyrase could have a potential role in human virulence (Fig. 6). Future research will help clarify the importance of P. falciparum E-NTPDase in the establishment of the severe form of malaria, parasite physiology, and signaling processes.

Proposed mechanism of action of E-NTPDase in P. falciparum. An enzyme able to hydrolyze ATP is present in the parasite external surface, which could convert ATP to AMP. The monophosphate nucleotide could then be converted into adenosine by a different type of nucleotidase. Since P. falciparum lacks the ability to synthesize the purine ring, the parasite relies on getting purine precursors, as adenosine, from the RBCs as an essential nutrient. Blocking this pathway impairs P. falciparum asexual development. The P. falciparum, E-NTPDase seems to be phylogenetically distinct from other parasites and it is not present in any other Plasmodium specie. Thus, we believe that this enzyme is also involved in the lethal form of the disease caused by P. falciparum. EM erythrocyte membrane, PVM parasitophorous vacuole membrane, PM parasite membrane
References
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI (2005) The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434(7030):214–217
Garcia CR, de Azevedo MF, Wunderlich G, Budu A, Young JA, Bannister L (2008) Plasmodium in the postgenomic era: new insights into the molecular cell biology of malaria parasites. Int Rev Cell Mol Biol 266:85–156
Wright GJ, Rayner JC (2014) Plasmodium falciparum erythrocyte invasion: combining function with immune evasion. PLoS Pathog 10(3):e1003943
Paloque L, Ramadani AP, Mercereau-Puijalon O, Augereau JM, Benoit-Vical F (2016) Plasmodium falciparum: multifaceted resistance to artemisinins. Malar J 15:149
Tumwebaze P, Tukwasibwe S, Taylor A, Conrad M, Ruhamyankaka E, Asua V, Walakira A, Nankabirwa J, Yeka A, Staedke SG, Greenhouse B, Nsobya SL, Kamya MR, Dorsey G, and Rosenthal PJ, Changing antimalarial drug resistance patterns identified by surveillance at three sites in Uganda. J Infect Dis, 2016
Sharma A, Santos IO, Gaur P, Ferreira VF, Garcia CR, da Rocha DR (2013) Addition of thiols to o-quinone methide: new 2-hydroxy-3-phenylsulfanylmethyl[1,4]naphthoquinones and their activity against the human malaria parasite Plasmodium falciparum (3D7). Eur J Med Chem 59:48–53
Budu A, Garcia CR (2012) Generation of second messengers in Plasmodium. Microbes Infect 14(10):787–795
Cruz LN, Wu Y, Ulrich H, Craig AG, Garcia CR (2016) Tumor necrosis factor reduces Plasmodium falciparum growth and activates calcium signaling in human malaria parasites. Biochim Biophys Acta 1860(7):1489–1497
McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung BK, Chatterjee AK, McCormack SL, Manary MJ, Zeeman AM, Dechering KJ, Kumar TR, Henrich PP, Gagaring K, Ibanez M, Kato N, Kuhen KL, Fischli C, Rottmann M, Plouffe DM, Bursulaya B, Meister S, Rameh L, Trappe J, Haasen D, Timmerman M, Sauerwein RW, Suwanarusk R, Russell B, Renia L, Nosten F, Tully DC, Kocken CH, Glynne RJ, Bodenreider C, Fidock DA, Diagana TT, Winzeler EA (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504(7479):248–253
Alam MM, Solyakov L, Bottrill AR, Flueck C, Siddiqui FA, Singh S, Mistry S, Viskaduraki M, Lee K, Hopp CS, Chitnis CE, Doerig C, Moon RW, Green JL, Holder AA, Baker DA, Tobin AB (2015) Phosphoproteomics reveals malaria parasite protein kinase G as a signalling hub regulating egress and invasion. Nat Commun 6:7285
Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedeberg's Arch Pharmacol 362(4–5):299–309
Fleck SL, Birdsall B, Babon J, Dluzewski AR, Martin SR, Morgan WD, Angov E, Kettleborough CA, Feeney J, Blackman MJ, Holder AA (2003) Suramin and suramin analogues inhibit merozoite surface protein-1 secondary processing and erythrocyte invasion by the malaria parasite Plasmodium falciparum. J Biol Chem 278(48):47670–47677
Levano-Garcia J, Dluzewski AR, Markus RP, Garcia CR (2010) Purinergic signalling is involved in the malaria parasite Plasmodium falciparum invasion to red blood cells. Purinergic Signal 6(4):365–372
Huber SM (2012) Purinoceptor signaling in malaria-infected erythrocytes. Microbes Infect 14(10):779–786
Cruz LN, Juliano MA, Budu A, Juliano L, Holder AA, Blackman MJ, Garcia CR (2012) Extracellular ATP triggers proteolysis and cytosolic Ca(2)(+) rise in plasmodium berghei and Plasmodium yoelii malaria parasites. Malar J 11:69
Santos RF, Possa MA, Bastos MS, Guedes PM, Almeida MR, Demarco R, Verjovski-Almeida S, Bahia MT, Fietto JL (2009) Influence of ecto-nucleoside triphosphate diphosphohydrolase activity on Trypanosoma cruzi infectivity and virulence. PLoS Negl Trop Dis 3(3):e387
Fietto JL, DeMarco R, Nascimento IP, Castro IM, Carvalho TM, de Souza W, Bahia MT, Alves MJ, Verjovski-Almeida S (2004) Characterization and immunolocalization of an NTP diphosphohydrolase of Trypanosoma cruzi. Biochem Biophys Res Commun 316(2):454–460
Asai T, Miura S, Sibley LD, Okabayashi H, Takeuchi T (1995) Biochemical and molecular characterization of nucleoside triphosphate hydrolase isozymes from the parasitic protozoan toxoplasma gondii. J Biol Chem 270(19):11391–11397
de Almeida Marques-da-Silva E, de Oliveira JC, Figueiredo AB, de Souza Lima Junior D, Carneiro CM, Rangel Fietto JL, Crocco Afonso LC (2008) Extracellular nucleotide metabolism in Leishmania: influence of adenosine in the establishment of infection. Microbes Infect 10(8):850–857
Gomes RS, de Carvalho LC, de Souza Vasconcellos R, Fietto JL, Afonso LC (2015) E-NTPDase (ecto-nucleoside triphosphate diphosphohydrolase) of Leishmania amazonensis inhibits macrophage activation. Microbes Infect 17(4):295–303
Vasconcellos Rde S, Mariotini-Moura C, Gomes RS, Serafim TD, Firmino Rde C, Silva EBM, Castro FF, Oliveira CM, Borges-Pereira L, de Souza AC, de Souza RF, Gomez GA, Pinheiro Ada C, Maciel TE, Silva-Junior A, Bressan GC, Almeida MR, Baqui MM, Afonso LC, Fietto JL (2014) Leishmania infantum ecto-nucleoside triphosphate diphosphohydrolase-2 is an apyrase involved in macrophage infection and expressed in infected dogs. PLoS Negl Trop Dis 8(11):e3309
Bisaggio DF, Peres-Sampaio CE, Meyer-Fernandes JR, Souto-Padron T (2003) Ecto-ATPase activity on the surface of Trypanosoma cruzi and its possible role in the parasite-host cell interaction. Parasitol Res 91(4):273–282
Silverman JA, Qi H, Riehl A, Beckers C, Nakaar V, Joiner KA (1998) Induced activation of the toxoplasma gondii nucleoside triphosphate hydrolase leads to depletion of host cell ATP levels and rapid exit of intracellular parasites from infected cells. J Biol Chem 273(20):12352–12359
Leite PM, Gomes RS, Figueiredo AB, Serafim TD, Tafuri WL, de Souza CC, Moura SA, Fietto JL, Melo MN, Ribeiro-Dias F, Oliveira MA, Rabello A, Afonso LC (2012) Ecto-nucleotidase activities of promastigotes from Leishmania (Viannia) braziliensis relates to parasite infectivity and disease clinical outcome. PLoS Negl Trop Dis 6(10):e1850
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B (2002) Genome sequence of the human malaria parasite plasmodium falciparum. Nature 419(6906):498–511
Sansom FM, Robson SC, Hartland EL (2008) Possible effects of microbial ecto-nucleoside triphosphate diphosphohydrolases on host-pathogen interactions. Microbiol Mol Biol Rev 72(4):765–781 Table of Contents
Trager W, Jensen JB (1976) Human malaria parasites in continuous culture. Science 193(4254):673–675
Lambros C, Vanderberg JP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65(3):418–420
Schuck DC, Ribeiro RY, Nery AA, Ulrich H, Garcia CR (2011) Flow cytometry as a tool for analyzing changes in Plasmodium falciparum cell cycle following treatment with indol compounds. Cytometry A 79(11):959–964
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Ekman P, Jager O (1993) Quantification of subnanomolar amounts of phosphate bound to seryl and threonyl residues in phosphoproteins using alkaline hydrolysis and malachite green. Anal Biochem 214(1):138–141
Wrenger C, Muller S (2004) The human malaria parasite Plasmodium falciparum has distinct organelle-specific lipoylation pathways. Mol Microbiol 53(1):103–113
Mariotini-Moura C, Bastos MS, de Castro FF, Trindade ML, de Souza Vasconcellos R, do Valle Neves MA, Moreira BP, de Freitas Santos R, de Oliveira CM, Cunha LC, Souto XM, Bressan GC, Silva-Junior A, Baqui MM, Bahia MT, de Almeida MR, Meyer-Fernandes JR, Fietto JL (2013) Trypanosoma cruzi nucleoside triphosphate diphosphohydrolase 1 (TcNTPDase-1) biochemical characterization, immunolocalization and possible role in host cell adhesion. Acta Trop 130C:140–147
de Souza MC, de Assis EA, Gomes RS, Marques A, da Silva E, Melo MN, Fietto JL, Afonso LC (2010) The influence of ecto-nucleotidases on Leishmania amazonensis infection and immune response in C57B/6 mice. Acta Trop 115(3):262–269
Marti E, Canti C, Gomez I, de Aranda, Miralles F, Solsona C (1996) Action of suramin upon ecto-apyrase activity and synaptic depression of torpedo electric organ. Br J Pharmacol 118(5):1232–1236
Escalada A, Navarro P, Ros E, Aleu J, Solsona C, Martin-Satue M (2004) Gadolinium inhibition of ecto-nucleoside triphosphate diphosphohydrolase activity in torpedo electric organ. Neurochem Res 29(9):1711–1714
Crack BE, Pollard CE, Beukers MW, Roberts SM, Hunt SF, Ingall AH, McKechnie KC, AP IJ, Leff P (1995) Pharmacological and biochemical analysis of FPL 67156, a novel, selective inhibitor of ecto-ATPase. Br J Pharmacol 114(2):475–481
Abraham EH, Sterling KM, Kim RJ, Salikhova AY, Huffman HB, Crockett MA, Johnston N, Parker HW, Boyle WE Jr, Hartov A, Demidenko E, Efird J, Kahn J, Grubman SA, Jefferson DM, Robson SC, Thakar JH, Lorico A, Rappa G, Sartorelli AC, Okunieff P (2001) Erythrocyte membrane ATP binding cassette (ABC) proteins: MRP1 and CFTR as well as CD39 (ecto-apyrase) involved in RBC ATP transport and elevated blood plasma ATP of cystic fibrosis. Blood Cells Mol Dis 27(1):165–180
Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ Jr, Treatman C, Wang H (2009) PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37(Database issue):D539–D543
Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA (2003) Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301(5639):1503–1508
Shock JL, Fischer KF, DeRisi JL (2007) Whole-genome analysis of mRNA decay in plasmodium falciparum reveals a global lengthening of mRNA half-life during the intra-erythrocytic development cycle. Genome Biol 8(7):R134
Zimmermann H, Zebisch M, Strater N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8(3):437–502
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4(6):1581–1590
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300(4):1005–1016
Pinheiro, C.M., E.S. Martins-Duarte, R.B. Ferraro, A.L. Fonseca de Souza, M.T. Gomes, A.H. Lopes, M.A. Vannier-Santos, A.L. Santos, and J.R. Meyer-Fernandes Leishmania amazonensis: Biological and biochemical characterization of ecto-nucleoside triphosphate diphosphohydrolase activities. Exp Parasitol, 2006. 114(1): p. 16–25.
Coimbra ES, Goncalves-da-Costa SC, Costa BL, Giarola NL, Rezende-Soares FA, Fessel MR, Ferreira AP, Souza CS, Abreu-Silva AL, Vasconcelos EG (2008) A Leishmania (L.) amazonensis ATP diphosphohydrolase isoform and potato apyrase share epitopes: antigenicity and correlation with disease progression. Parasitology 135(3):327–335
Asai T, O'Sullivan WJ, Tatibana M (1983) A potent nucleoside triphosphate hydrolase from the parasitic protozoan Toxoplasma gondii. Purification, some properties, and activation by thiol compounds. J Biol Chem 258(11):6816–6822
Sibley LD, Niesman IR, Asai T, Takeuchi T (1994) Toxoplasma gondii: secretion of a potent nucleoside triphosphate hydrolase into the parasitophorous vacuole. Exp Parasitol 79(3):301–311
Sansom FM (2012) The role of the NTPDase enzyme family in parasites: what do we know, and where to from here? Parasitology 139(8):963–980
Sevigny J, Martin-Satue M, Pintor J (2015) Purinergic signalling in immune system regulation in health and disease. Mediat Inflamm 2015:106863
Frech C, Chen N (2011) Genome comparison of human and non-human malaria parasites reveals species subset-specific genes potentially linked to human disease. PLoS Comput Biol 7(12):e1002320
Sansom FM, Riedmaier P, Newton HJ, Dunstone MA, Muller CE, Stephan H, Byres E, Beddoe T, Rossjohn J, Cowan PJ, d'Apice AJ, Robson SC, Hartland EL (2008) Enzymatic properties of an ecto-nucleoside triphosphate diphosphohydrolase from Legionella pneumophila: substrate specificity and requirement for virulence. J Biol Chem 283(19):12909–12918
Wang CJ, Vlajkovic SM, Housley GD, Braun N, Zimmermann H, Robson SC, Sevigny J, Soeller C, Thorne PR (2005) C-terminal splicing of NTPDase2 provides distinctive catalytic properties, cellular distribution and enzyme regulation. Biochem J 385(Pt 3):729–736
Acknowledgments
The authors thank Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for the financial support and COLSAN for providing the blood and plasma.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with participants or animals performed by any of the authors.
Funding
This work was supported by grants from FAPESP (Process 11/51295–5 to CRSG, 2013/10288-1 and 2015/26722-8 to CW, 2012/12807-3 to KAM and 2010/51593-3 to LBP).
Conflicts of interest
Lucas Borges-Pereira declares that he has no conflict of interest. Kamila Anna Meissner declares that she has no conflict of interest. Carsten Wrenger declares that he has no conflict of interest. Célia R. S. Garcia declares that she has no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Borges-Pereira, L., Meissner, K.A., Wrenger, C. et al. Plasmodium falciparum GFP-E-NTPDase expression at the intraerythrocytic stages and its inhibition blocks the development of the human malaria parasite. Purinergic Signalling 13, 267–277 (2017). https://doi.org/10.1007/s11302-017-9557-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-017-9557-4