
Abstract
A better understanding of the physiological effects of guanosine-based purines should help clarify the complex subject of purinergic signalling. We studied the effect of extracellular guanosine 5′triphosphate (GTP) on the differentiation of two excitable cell lines that both have specific binding sites for GTP: PC12 rat pheochromocytoma cells and C2C12 mouse skeletal muscle cells. PC12 cells can be differentiated into fully functional sympathetic-like neurons with 50′00 ng ml−1 of nerve growth factor, whereas serum starvation causes C2C12 cells to differentiate into myotubes showing functional excitation–contraction coupling, with the expression of myosin heavy chain proteins. Our results show that GTP enhances the differentiation of both of these excitable cell lines. The early events in guanosine-based purine signal transduction appear to involve an increase in intracellular Ca2+ levels and membrane hyperpolarization. We further investigated the early activation of extracellular-regulated kinases and phosphoinositide 3-kinase in GTP-stimulated PC12 and C2C12 cells, respectively. We found that GTP promotes the activation of both kinases. Together, our results suggest that, even if there are some differences in the signalling pathways, GTP-induced differentiation in both cell lines is dependent on an increase in intracellular Ca2+.
Similar content being viewed by others

Introduction
Extracellular purines exert a variety of physiological functions that influence cell growth, differentiation, and death [1]. Although a biological role of adenosine-based purines in the peripheral and central nervous system was identified several years ago [2], the physiological role of guanosine-based purines has only recently been established [1, 3]. Guanosine and its nucleotide, in particular guanosine 5′ triphosphate (GTP), were found to play a role in synaptic transmission. In fact, GTP, like ATP, is stored in synaptic vesicles and co-released with neurotransmitters in the synaptic cleft [4]. In addition, GTP, as well as guanosine, released from neurons and astrocytes has not only a physiological role but is also involved in pathological conditions [5]. In this scenario it is interesting to note that extracellular GTP and guanosine are able to act as trophic and mitogenic factors in neuronal and glial compartments [1, 6, 7]. In fact, GTP is able to modulate glial proliferation and neuron differentiation at the same extent or even more significantly than the adenine-based nucleotides do [7, 8].
Other excitable tissues, including skeletal muscle cells, are sensitive to GTP. In fact, extracellular GTP has been shown to modulate the development of isometric twitch tension in frog muscle fibres [9]. Moreover, extracellular purines affect the behaviour of Paramecium tetraurelia and Tetrahymena thermophila, inducing backward swimming [10]. These ciliates are good eukaryotic sensory models in which to study purinergic signalling. They present membrane binding sites to purine showing physiological mechanisms of adaptation [11, 12]. Extracellular micromolar concentrations of GTP induced cell membrane depolarization and activate cellular mechanisms of retraction [13, 14]. It was speculated that this behaviour is a kind of defence mechanism, since GTP or ATP are not toxic to these cells, but their high concentrations could be interpreted as the result of cell lyses consequent from the presence of dangerous conditions [10].
We studied the effects of extracellular GTP in PC12 and C2C12 cells, which are models of neuronal and skeletal muscle cells, respectively [15, 16]. PC12 cells are derived from a rat pheochromocytoma and differentiate in the presence of nerve growth factor (NGF; 50–100 ng ml−1) into a fully functional sympathetic (adrenergic) neuronal phenotype within 7 days [17]. C2C12 cells are satellite cells established by subcloning the C2 cell line [18], which was originally derived from the thigh muscle of a 2-month old mouse [19]. This diploid continuous cell line is frequently used as a model to study skeletal muscle differentiation and function.
In this mini-review we report our studies on the role of GTP in the differentiation of two excitable cell lines, PC12 and C2C12. Following our previous results showing that these cells have specific binding sites for GTP [15, 16], we focused our attention on the biological effects and signal transduction that GTP induces in these cell lines.
In particular, we examined the effects of GTP on neurite outgrowth in PC12 cells in the presence or absence of differentiation-inducing concentrations of NGF. On the other hand, we also investigated the effect of GTP on skeletal muscle differentiation, analysing the expression of myosin heavy chain (MyHC) protein in C2C12 cells following serum starvation-induced differentiation. Considering the known specific intracellular pathways activated during the differentiation processes, we examined the role of extracellular regulated kinases (ERKs) and phosphoinositide 3-kinase (PI3K) in the effects of GTP on these differentiating cell lines.
GTP effects on differentiation of excitable cells
In our previous studies we examined the binding of GTP to the plasma membrane of C2C12 and PC12 cells. On intact C2C12 cells we found two specific binding sites with distinct characteristics: high-affinity sites with a Kd of 37.2 ± 13.7 µM and a Bmax of 2.6 ± 0.3 nmol mg−1 protein, and low-affinity sites with a Kd of 186.5 ± 96.0 µM and a Bmax of 22.3 ± 3.1 nmol mg−1 protein. On PC12 cells, we found only one class of specific binding sites, with a Kd of 53.9 ± 10 µM and a Bmax of 2.15 ± 0.12 µM. In both cell lines, ATP and UTP were unable to displace or compete with GTP binding [15, 16].
We expected that the early events in GTP signal transduction would include an increase in [Ca2+]i. In PC12 cells, GTP induces a slight influx of calcium through L-type calcium channels, resulting in a robust and long-term increase in [Ca2+]i due to release of intracellular calcium from ryanodine-sensitive stores [20]. The GTPinduced [Ca2+]i increase had different kinetics in C2C12 cells than in PC12 cells. Specifically, GTP-stimulated C2C12 cells showed an increase in [Ca2+]i primarily due to release from intracellular stores [16].
Using purinoceptor antagonists (suramin, Basilen Blue, PPDAS, and NF023), we established that, on both cell lines, GTP activates a receptor belonging to the P2 class and, in particular, a metabotropic-like one [15, 16, 20]. In PC12 cells GTP enhances protein kinase C activity, suggesting the involvement of phospholipase C in the nucleotide signal transductive mechanism [15]. Moreover, overnight incubation of PC12 cells with pertussis toxin, an inactivator of Gi/0 proteins, reduced their response to GTP, confirming the participation of a P2-class receptor in the effects of GTP [20].
In C2C12 cell line, other evidence showed the metabotropic feature of the GTP-activated receptor. In fact, suramin and Basilen Blue prevented GTP binding to the C2C12 membranes and blocked GTP-induced [Ca2+]i rise [16]. In addition, this last GTP-induced effect is mediated by calcium release from the internal store, a mechanism triggered by phospholipase C activity and inositol triphosphate (IP3) and diacylglycerol (DAG) production, usually due to metabotropic purinoceptor activation in these cells [21].
However, in C12C12 myoblasts, there was also a component of Ca2+ influx in the GTP-induced increase in [Ca2+]i. Therefore, given their similar Kd values, we predict that the sites on C2C12 and PC12 cells have common properties.
Because of the involvement of [Ca2+]i in the electrophysiological properties of the plasma membrane in cells differentiating towards an excitable phenotype, we investigated whether the GTP-induced increase of [Ca2+]i altered the membrane polarization in single cells. We carried out video-imaging analysis of cells incubated with Fura and DiBAC4, which are fluorescent dyes used to monitor [Ca2+]i and membrane polarization, respectively [22]. We found that GTP caused an increase in [Ca2+]i and, only a few seconds later, hyperpolarization of the plasma membrane in both PC12 and C2C12 cells [20, 23].
Based on the results of these experiments, we hypothesized that the observed hyperpolarization in both cell types is due to a Ca2+-induced outward K+ current. Indeed, emptying the Ca2+ stores in C2C12 cells with thapsigargin or blocking calcium influx in PC12 cells with nifedipine eliminated the GTP-induced increase in [Ca2+]i as well as the K+ current. Moreover, 10 µM clotrimazole, a specific inhibitor of the Ca2+-activated K+ channel, blocked GTP-dependent hyperpolarization [20, 23].
However, the GTP-dependent hyperpolarization via Ca2+-activated K+ channel modulation probably had a complex role. In PC12 cells Ca2+-activated K+ channel modulation probably had little trophic importance because inhibiting the GTP-dependent hyperpolarization with clotrimazole did not prevent GTP-induced neurite outgrowth [20]. On the contrary, the pharmacological blockade of membrane hyperpolarization prevented the ability of low- and high-affinity GTP binding sites to activate C2C12 differentiation [23].
Under the appropriate environmental conditions, C2C12 and PC12 cell lines can be differentiated into excitable phenotypes, namely, muscle and neuron, respectively. The differentiation of C2C12 cells is induced by simple serum starvation and is characterized by proliferation prior to the establishment of a post-mitotic state, followed by the expression of MyHC proteins and cell fusion [24]. The differentiation of PC12 cells into a sympathetic phenotype requires the presence of a specific differentiating factor, namely NGF [17].
We next investigated the role of GTP on both NGF-induced differentiation of PC12 cells and serum starvation-induced differentiation of C2C12 cells. We assessed the differentiation in these cells by measuring two specific markers: neurite outgrowth in PC12 cells and MyHC protein expression in C2C12 cells. We found that GTP enhances NGF-induced neurite outgrowth in PC12 cells and that it increases the amount of MyHC proteins after cell proliferative boost in differentiating C2C12 cells. In both cellular models these effects were blocked by metabotropic purinoceptor antagonists as well as those used pharmacologically to inhibit the GTP-induced increase in [Ca2+]i [20, 23].
We further examined the signalling events involved in the early stages of GTP-induced differentiation. We primarily considered the possible GTP modulation of well-known key steps in PC12 and C2C12 differentiation without groping in the complex intracellular signalling pathways.
In PC12 cells, activation of ERK1/2 is known to participate in the initial phases of NGF-induced differentiation by activating the expression of early genes involved in cell cycle arrest and differentiation [25]. We therefore examined the phosphorylation of ERK1/2 in PC12 cells stimulated with NGF in the presence or absence of GTP. We found that extracellular GTP caused a relatively longterm increase in NGF-induced phosphorylation of ERK1/2 [20].
PI3Ks, a family of lipid kinases that induce signals by phosphorylating the hydroxyl group at the 3-position of phosphoinositides, are directly involved in the process of myogenesis. PI3Ks regulate a number of physiological functions, including membrane trafficking, cell adhesion, actin rearrangement, and cell growth [26]. Activation of PI3Ks occurs downstream of both G-protein-coupled receptors and receptor tyrosine kinases (e.g., insulin and insulin-like growth factor I receptors). In particular, PI3Ks are required for myotube formation in C2C12 cells [27]. To examine the role of PI3Ks in GTP-induced muscle differentiation, we examined the effect of the PI3K inhibitor LY294002. This compound prevented an otherwise effective concentration of GTP from increasing the fusion index of MyHC-positive C2C12 [28]. This result supports the notion that PI3K is involved in GTP-induced differentiation of C2C12 cells.
Conclusions and perspectives
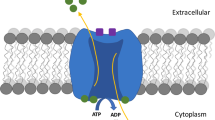
Based on the results presented here, we developed the following model for the common GTP-induced signalling events (Scheme 1).

Scheme of the GTP signal transduction pathway in PC12 and C2C12 cells. RyR ryanodine receptor, IP 3 inositol triphosphate, G G-protein, PLC phospholipase C, DA diacylglycerol
As shown in the scheme, on both neuronal-like PC12 and muscle-like C2C12 cells, extracellular GTP binds to specific, P2Y receptor-like sites. This induces an increase in [Ca2+]i that, in turn, causes membrane hyperpolarization through K+ channels. However, the kinetics and the source of the Ca2+ are different in the two cell lines. In particular, in PC12 cells, the [Ca2+]i increase is derived from an initial influx of Ca2+ from the extracellular compartment, which induces Ca2+ release from intracellular ryanodine-sensitive stores. This leads to the activation of ERKs, which enhances neuronal differentiation. In C2C12 cells, effective concentrations of extracellular GTP provoke an increase in [Ca2+]i due to release from intracellular IP3-sensitive stores. This enhances the expression of MyHC in C2C12 myoblasts and commits them to fuse into multinucleated myotubes, probably via a PI3K-dependent signal transduction mechanism.
Considering that this study is far from depicting the complete puzzle of GTP signal transduction pathways and that a lot of upstream and downstream steps between [Ca2+]i rise and ERK or PI3K regulation should be investigated, our in vitro models present interesting plasticity and easy handling for further in-depth studies. At present, our attention is also focused on two aims. One concerns the potential role of GTP as intercellular signal between neuron and skeletal muscle fibres, not only in their synaptic transmission but also as a regulating trophic factor. The other aim, on the other hand, investigates whether GTP is able to influence nuclear activity and modulate specific target genes. This could confirm the functions of GTP, not only as an extracellular molecule regulating cytoplasmic activity and cell adaptation, but also as a full trophic factor.
Abbreviations
- [Ca2+]i :
-
intracellular Ca2+ concentration
- DAG:
-
diacylglycerol
- DM:
-
differentiating medium
- ERK:
-
extracellular regulated kinase
- GTP:
-
guanosine 5′ triphosphate
- IP3:
-
inositol triphosphate
- MyHC:
-
myosin heavy chain
- NGF:
-
nerve growth factor
- PI3K:
-
phosphoinositide 3-kinase
References
Rathbone MP, Middlemiss PJ, Gysbers JW et al (1999) Trophic effects of purines in neurons and glial cells. Prog Neurobiol 59:663′90
Burnstock G (1997) The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology 36:1127′139
Franke H, Illes P (2006) Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol Ther 109:297′24
Wagner JA, Carlson SS, Kelly RB (1978) Chemical and physical characterization of cholinergic synaptic vesicles. Biochemistry 17:1199′206
Ciccarelli R, Di Iorio P, Giuliani P et al (1999) Rat cultured astrocytes release guanine-based purines in basal conditions and after hypoxia/hypoglycemia. Glia 25:93′8
Neary JT, Rathbone MP, Cattabeni F et al (1996) Trophic actions of extracellular nucleotides and nucleosides on glial and neuronal cells. Trends Neurosci 19:13′8
Rathbone MP, Middlemiss PJ, Gysbers JW et al (1992) Purine nucleosides and nucleotides stimulate proliferation of a wide range of cell types. In vitro Cell Dev Biol 28A:529′36
Gysbers JW, Rathbone MP (1996) GTP and guanosine synergistically enhance NGF-induced neurite outgrowth from PC12 cells. Int J Dev Neurosci 14:19′4
Mancinelli L, Fanò G, Ferroni L et al (1983) Evidence for an ionotropic positive action of cGMP during excitation-contraction coupling in frog sartorius muscle. Can J Physiol Pharm 61:590′94
Hennessey TM (2005) Responses of the ciliates Tetrahymena and Paramecium to external ATP and GTP. Purinergic Signalling 1:101′10
Kim MY, Kuruvilla HG, Raghu S, Hennessey TM (1999) ATP reception and chemosensory adaptation in Tetrahymena thermophila. J Exp Biol 202:407′16
Kim MY, Kuruvilla HG, Hennessey TM (1997) Chemosensory adaptation in Paramecium involves changes in both repellent binding and the consequent receptor potentials. J Comp Biochem Physiol 118:589′97
Clark KD, Hennessey TM, Nelson DL et al (1997) Extracellular GTP causes membrane-potential oscillations through the parallel activation of Mg2+ and Na+ currents in Paramecium tetraurelia. Biology 157:159′67
Mimikakis JL, Nelson DL (1998) Evidence for two separate purinergic responses in Paramecium tetraurelia: XTP inhibits only the oscillatory responses to GTP. J Membr Biol 163:19′3
Gysbers JW, Guarnieri S, Mariggiò MA et al (2000) Extracellular guanosine 5′triphosphate enhances nerve growth factor-induced neurite outgrowth via increases in intracellular calcium. Neuroscience 96:817′24
Pietrangelo T, Mariggiò MA, Lorenzon P et al (2002) Characterization of specific GTP binding sites in C2C12 mouse skeletal muscle cells. J Muscle Res Cell Motil 23:107′18
Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73:2424′428
Blau HM, Webster C, Pavlath GK (1983) Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 80:4856′860
Yaffe D, Saxel O (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270:725′27
Guarnieri S, Fanò G, Rathbone MP, Mariggiò MA (2004) Cooperation in signal transduction of extracellular guanosine 5′triphosphate and nerve growth factor in neuronal differentiation of PC12 cells. Neuroscience 128:697′12
Henning RH, Duin M, den Hertog A, Nelemans A (1993) Activation of the phospholipase C pathway by ATP is mediated exclusively through nucleotide type P2-purinoceptors in C2C12 myotubes. Br J Pharmacol 110(2):747′52
Gonzalez JE, Tsien RY (1995) Voltage sensing by fluorescence resonance energy transfer in single cells. Biophys J 4:1272′280
Pietrangelo T, Fioretti B, Mancinelli R et al (2006) Extracellular guanosine-5′triphosphate modulates myogenesis via intermediate Ca2+-activated K+ currents on C2C12 mouse cells. J Physiol 572:721′33
Andres V, Walsh K (1996) Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol 132:657′66
Vaudry D, Stork PJ, Lazarovici P et al (2002) Signaling pathways for PC12 cell differentiation: making the right connections. Science 296:1648′649
Toker A, Cantley LC (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387:673′76
Sumitani S, Goya K, Testa JR et al (2002) Akt1 and Akt2 differently regulate muscle creatine kinase and myogenin gene transcription in insulin-induced differentiation of C2C12 myoblasts. Endocrinology 143:820′28
Pietrangelo T, Mancinelli R, Fanò G, Fulle S (2005) Extracellular GTP modulates the genic activation during myogenic differentiation process. Proceedings of the National Conference of Italian Physiological Society SIF Palermo, Italy
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pietrangelo, T., Guarnieri, S., Fulle, S. et al. Signal transduction events induced by extracellular guanosine 5′triphosphate in excitable cells. Purinergic Signalling 2, 633–636 (2006). https://doi.org/10.1007/s11302-006-9021-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-006-9021-3