
Abstract
Influenza A virus infection induces type I interferons (IFNs α/β) which activate host antiviral responses through a cascade of IFN signaling events. Herein, we compared highly pathogenic H5N1 and low pathogenic H11N1 avian influenza viruses isolated from India, for their replication kinetics and ability to induce IFN-β and interferon-stimulating genes (ISGs). The H5N1 virus showed a higher replication rate and induced less IFN-β and ISGs compared to the H11N1 virus when grown in the human lung epithelial A549 cells, reflecting the generation of differential innate immune responses during infection by these viruses. The non-structural 1 (NS1) protein, a major IFN-antagonist, known to help the virus in evading host innate immune response was compared from both the strains using bioinformatics tools. Analyses revealed differences in the composition of the NS1 proteins from the two strains that may have an impact on the modulation of the innate immune response. Intriguingly, H5N1 virus attenuated IFN-β response in a non-NS1 manner, suggesting the possible involvement of other viral proteins (PB2, PA, PB1/PB1-F2) of H5N1 in synergy with NS1. Preliminary analyses of the above proteins of the two strains by sequence comparison show differences in charged residues. The insight gained will be useful in designing experimental studies to elucidate a probable role of the polymerase protein(s) in association with NS1 in inhibiting the IFN signaling and understanding the molecular mechanism governing the difference.
Similar content being viewed by others

Introduction
Influenza A viruses (IAV) of the family Orthomyxoviridae are zoonotic pathogens which pose a serious threat to human health globally. Because of their ability to reassort, they adapt to new hosts ensuring the generation of newer strains with varied degrees of pathogenicity and transmissibility [1, 2]. Avian influenza (AI) viruses cause infections in poultry, and wild and migratory birds with sporadic bird-to-human transmission [3,4,5]. Aquatic birds are the natural reservoirs [6] and to date all the AI subtypes have been isolated from them, except for the new bat-origin H17N10 and H18N11 [2]. AI viruses are classified as highly pathogenic avian influenza (HPAI) and low pathogenic avian influenza (LPAI) viruses. LPAI viruses are believed to be the genesis of HPAI viruses [3, 7]. Since the detection and isolation of HPAI–H5N1 from China in 1996 [8], the virus has spread in Asia causing outbreaks in poultry and human zoonotic infections [4, 9]. India has witnessed several outbreaks of the HPAI–H5N1 strain in domestic poultry since 2006 [10,11,12], which has prompted AI surveillance studies at the animal–human interface to understand the role of birds in the transmission of AI viruses.
The host innate immune responses serve as the first line of defense during viral infection restricting viral replication and spread. Type I interferons (IFNs) represented by IFN-α/β are secreted as host antiviral responses during IAV infection sensed by Retinoic acid-like receptor (RLR) genes and expression of the IFN-stimulated genes (ISGs) through the JAK-STAT signaling pathway [13,14,15]. Non-structural protein-1 (NS1) acts as a principal IFN-β antagonist by interfering with IFN production [14, 16] and IAVs lacking the NS1 gene can replicate in an IFN-deficient system [17], suggesting that HPAI strains may express NS1 that suppresses the host IFN-mediated antiviral effects. This premise set a platform to study differences in the host IFN-mediated responses elicited towards HPAI and LPAI viruses.
In the present study, we selected two unique AI strains, HPAI–H5N1 and LPAI–H11N1, that differ in their genetic organization and pathogenicity [11, 18, 19], to study the host-mediated IFN responses and their modulation in human lung epithelial A549 cells. The HPAI–H5N1 virus isolated from the Manipur outbreak in 2007 was unrelated to the earlier H5N1 viruses in India [11]. The chosen LPAI–H11N1 virus was a reassortant isolated for the first time in India from a wild aquatic bird, with internal genes (NP, M) having 100% amino acid identity with the H5N1 virus [18]. Moreover, a microarray-based study reported that the H11N1–NS1 has the potential to modulate host–cellular environment [20]. Thus, we compared the replication kinetics of these viruses, gene-expression profile of IFN-β and ISGs, and the ability of the respective NS1 to inhibit the IFN-β responses. Using bioinformatics tools (sequence alignment and 3D structure prediction), we also compared the NS1 proteins.
Materials and methods
Viruses and cells
The viruses used in this study were isolated from India, namely A/Ck/India/NIV9743/07 (H5N1) [11] and A/Aquaticbird/India/NIV-17095/2007 (H11N1) [18]. The preparation of the virus stocks, hemagglutination (HA) assay was essentially as described earlier [21]. The HA titer for both the viruses was 512 HA units. The human lung epithelial A549 cell line used for all infection studies was maintained in Dulbecco’s modified Eagle medium (DMEM; Invitrogen Life Technologies, USA) with 10% fetal bovine serum (FBS, Invitrogen), 100 U/ml penicillin, and 100 µg/ml streptomycin. Madin–Darby Canine Kidney (MDCK) cells were obtained from the American Type Culture Collection (ATCC), USA, and used for determining the plaque-forming unit (pfu) by the standard plaque assays as a measure of the infectious virus titer of the stock as well as the infected cells for growth kinetic studies. MDCK cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin in tissue culture flasks at 37 °C in 5% CO2.
Plaque assay
Madin–Darby Canine Kidney (MDCK) cells were seeded at a cell density of 0.7 × 106 cells per well in a six-well tissue culture plate (Nunc, Denmark) and grown at 37 °C in a CO2 incubator. After the cells attained confluency, the confluent monolayers were washed with DMEM medium without FBS and infected with serial virus dilutions (10−1 till 10−5) and incubated for 1 h at 37 °C. After the incubation period, the cells were overlaid with overlay media containing 0.18% SeaKem LE Agarose (Lonza, Switzerland) and allowed to solidify. After 48 h of incubation at 37 °C, the plaques were stained with 0.1% crystal violet and counted. The virus titer was calculated as plaque-forming unit per milliliter (PFU/ml).
Viral replication experiments
Growth kinetics of both the viruses (H5N1 and H11N1) was monitored in A549 cells and the titers determined by plaque assay in MDCK cells. In brief, A549 cells (1.5 × 105 cells) were infected at a multiplicity of infection (MOI) of 0.01 with the viruses independently and incubated for the desired time. The medium for H11N1 virus contained 2 µg/ml Tosyl phenylalanyl chloromethyl ketone (TPCK) trypsin. Supernatants were collected at various time points of incubation such as 2, 12, 24, 48, and 72 h. The viral titer of the infected supernatants of all the above time points was measured by determining the pfus by the standard plaque assay in MDCK cells. The experiment was conducted thrice independently. Infection experiments with HPAI–H5N1 virus were conducted in a Biosafety level 3 (BSL3) laboratory.
Real-time PCR experiments
Innate immune response to these viruses and the M gene content was studied by quantitative gene-expression profiling using Real-time RT-PCR. In brief, A549 cells were infected with the respective viruses at MOI 4. RNA was isolated from the infected cells at 12 h post-infection (hpi) using the RNeasy mini kit (Qiagen, Germany). DNA traces were removed by DNase I (Promega, USA) treatment. The respective RNAs along with controls (636 ng) were subjected to cDNA preparation using High-Capacity cDNA Reverse Transcription Kit (ABI, USA). Innate immune response genes and the M gene content were quantitated by SYBR green real-time RT-PCR (ABI) in ABI 7500 fast real-time PCR system and normalized to the levels of 18S mRNA. The PCR conditions were as follows: 95 °C for 10 min followed by 40 cycles of 94 °C for 15 s, 55 °C for 60 s, and 72 °C for 60 s. Primer sequences of the target genes are listed in Table 1. The data are presented as relative mRNA expression fold induction and are representative of two experiments of three independent replicates. The relative expression levels of the target genes were analyzed by the comparative ΔΔCt method [22].
Luciferase assays
NS1 genes from the H11N1 and H5N1 viruses were amplified by RT-PCR and cloned into the mammalian expression vector pCDNA3.1 (Invitrogen). The reporter plasmid expressing the firefly luciferase under the control of IFN-β promoter (pGL4-IFN-Luc) was generated by cloning the human IFN-β promoter amplified from human lymphocyte RNA by RT-PCR into pGL4 (Promega) vector upstream of the luciferase gene. Transient transfection was performed in subconfluent A549 cells in 12-well cell culture plates. In brief, 500 ng of pGL4-IFN-β-Luc reporter plasmid, 2 or 3 µg of either pCDNA-H11N1–NS1 or pCDNA-H5N1-NS1 expression plasmid, and 5 ng of Renilla luciferase plasmid as an internal control for normalization of transfection efficiency were mixed with Lipofectamine 2000 transfection reagent (Invitrogen) and incubated for 20 min at room temperature (RT) before adding to the cells. Sixteen hours post-transfection, cells were induced by adding 500 ng of PolyI:C (InvivoGen, USA) as extracellular stimulation. Cells were analyzed for reporter activation 40 h post-transfection by measuring the firefly luciferase activity and the Renilla luciferase activity using the Dual-Glo Luciferase Assay System (Promega), according to the manufacturer’s protocol. The luminescence was read by a VICTOR X3 Multilabel Plate Reader (Perkin Elmer). Respective controls such as mock (lacking NS1 plasmid and PolyI:C) and a positive control (PC; lacking only NS1 expression plasmid) were used for comparison.
Quantitation of IFN-β by ELISA
A549 cells were infected with the H5N1 and H11N1 viruses at MOI 4 in a 6-well cell culture plate. Mock-infected cells received an equal amount of phosphate-buffered saline (PBS), pH 7.4. Cells were incubated at 37 °C for 24 h in a CO2 incubator. After 24 hpi, 100 µl of supernatant from the mock-infected as well as the virus-infected cells were added to a 96-well plate for determination of IFN-β concentration. ELISA was performed along with the standards according to the manufacturer’s protocol (PBL Assay Science, USA). The absorbance was measured at 450 nm. The amount of IFN-β was estimated from the standard curve. Statistical significance was determined by the Student’s t test.
Western blot and quantitation
A549 cells were infected with the H11N1 and H5N1 viruses at MOI 4. Total cellular protein from the infected cells was extracted using cell lysis buffer (1% NP40) 24 h post-infection. Protein content of the cell lysates was determined using the Bradford reagent (Biorad, USA). Equal amount of protein (25 μg) was separated on 12% SDS-PAGE and transferred to PVDF membrane (Amersham Bioscience, USA) at 100 V for 1 h. After blocking, the membrane was probed with a monoclonal anti-NS1 antibody (Santa Cruz Biotechnology, USA). For Western blots, the proteins were visualized by chemiluminescence using the ECL detection system (Amersham Bioscience). The blot was stripped and re-probed with anti β-actin antibody (Santa Cruz Biotechnology) as the loading control. For quantification of the protein content, scanned images of the Western blot were analyzed by densitometry using ImageJ (Image Processing and Analysis in Java, National Institute of Health, USA) software.
Bioinformatics analyses
In order to understand the differences between the NS1 proteins from the two strains at the molecular level, sequence analyses and structural comparison were carried out using bioinformatics tools. The amino acid sequences of NS1 protein from the H5N1 and H11N1 viruses were aligned using FASTA software (https://fasta.bioch.virginia.edu/). The 3D structures for these sequences were predicted by an homology-based modeling protocol as implemented in the SWISS-MODEL online server (https://swissmodel.expasy.org/) with the automated mode. The predicted structures were evaluated using PROCHECK analysis (http://www.ebi.ac.uk/thorntonsrv/software/PROCHECK). Minimized energy for the structures was calculated using GROMOS96 force-field in SwissPDBViewer (SPDBV) [23]. Visualization of the structures was carried out using Discovery Studio Viewer 4.1. Surface electrostatic analysis for the structures was carried out in NOC software v3.01 [24].
Statistical analysis
The Student’s t test was applied for the replication kinetics, comparison of the M gene content and the innate immune response genes, luciferase assays, and IFN-β content.
Results and discussion
Growth kinetics of HPAI–H5N1 and LPAI–H11N1 strains
To ascertain viable virus production, first we compared the growth kinetics of the HPAI and LPAI viruses-A/Ck/India/NIV9743/07 (H5N1) [11] and A/Aquaticbird/India/NIV-17095/2007 (H11N1) [18] in the human lung A549 cells. Virus stock preparation and hemagglutination (HA) assay were performed as described earlier [21]. The infectious virus titer of both the viruses was estimated by determining the pfu by the standard plaque assay. A549 cells were infected with the LPAI and HPAI viruses at equal multiplicity of infection (MOI) 0.01 and the infectious virus titers (pfu) were analyzed as a function of time by the standard plaque assay. Both the isolates replicated well in A549 cells. The virus titers between the two isolates differed significantly (P < 0.01) over time after 12 h until 24 h, and remained as a plateau until 72 h (Fig. 1a). It is also clear from the figure that the difference in the infectious virus titer between these isolates remained the same until 48 hpi. These data suggest that there is no difference in the early stages of infection between the viruses; however after 12 hpi, the higher replication rate of H5N1 compared to H11N1 virus results in higher titers. Further insight regarding the replication potential of the viruses was confirmed by an immunofluorescence assay using the anti-NP antibody (Fig. S1). The figure clearly shows an increased number of NP-expressed cells in both the HPAI and LPAI viruses at the two time points.

Replication kinetics of the H11N1 and H5N1 viruses in A549 cells and differential expression of innate immune response genes. a The viral titer of the infected supernatants of A549 cells at timepoints (2, 12, 24, 48, and 72 h) was determined by plaque assay in MDCK cells. The graphs represent mean ± SEM of three independent experiments. Asterisks indicate the statistical significance between H11N1 and H5N1 viruses (**P < 0.01; Student’s t test). b M gene expression in H11N1 and H5N1 virus-infected A549 cells and c–i Innate immune response against H11N1 and H5N1 in A549 cells by quantitative SYBR green real-time RT-PCR. Results were analyzed by the comparative ΔΔCt method. Asterisks indicate the statistical significance between mRNA expressions of the H5N1 and H11N1 viruses by Student’s t test (*P < 0.1; **P < 0.05; ***P < 0.01)
Gene-expression profile of innate immune response genes
Next, we sought to determine whether the above difference in the replication rate is due to the viruses’ ability to initiate type I IFN responses in A549-infected cells (MOI 4), by studying gene-expression profiling of the innate immune response and pattern-recognition receptor genes, and the M gene content (Table 1) using quantitative SYBR green real-time RT-PCR. The M gene content (Fig. 1b) was found to be consistent with the replication pattern of the respective viruses (Fig. 1a). We observed that the expression of IFN-β and RLR genes (RIG-1 and MDA-5) was significantly higher (P < 0.05) in the H11N1 virus-infected cells compared to the H5N1 virus (Fig. 1c–e). There was a marginal increase in RIG-1 expression and significantly higher (P < 0.01) expression of MDA-5 in the H11N1 virus-infected cells. Among the ISG’s analyzed in this study, expression of ISG-15 was marginally increased, whereas the expression of IFITM, MX1, and PKR genes was significantly (P < 0.05) higher in the H11N1 virus-infected cells than the H5N1 virus-infected cells (Fig. 1f–i). These results clearly indicate an increased amount of the intracellular RNA sensors in the LPAI virus-infected cells that probably enhance the expression of host IFN-β and ISG’s. Consistent with the elevated level of the IFN-β gene, higher amounts of IFN-β was expressed by the LPAI–H11N1 compared to the HPAI–H5N1 virus as determined by ELISA (Fig. 2). Similar studies with HPAI–H5N1 and the prototypic seasonal H3N2 strain suggest that differential IFN activation depends on the RIG-1 gene [25].

Quantitation of IFN-β from H5N1 virus- and H11N1 virus-infected cells by ELISA. A549 cells were either infected with the H5N1 and H11N1 viruses at MOI 4 or mock-infected and incubated at 37 °C for 24 h. IFN-β concentration in the cell supernatants was determined by ELISA after 24 h. Asterisk indicates statistical significance by the Student’s t test (*P < 0.05)
Effect of NS1 on IFN-β expression
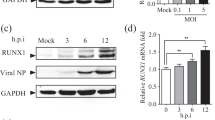
Subsequently, we asked whether the lower expression of IFN-β during H5N1-virus infection compared to H11N1-virus infection was due to the inhibitory action of NS1 on the production of IFN-β, by comparing the capacity of transiently expressed NS1 of the respective strains for inhibiting the IFN-β promoter in A549 cells using the Luciferase reporter assay system. Normalization of the transfection efficiency was achieved by cotransfecting the Renilla luciferase plasmid as an internal control. Results displayed in Fig. 3a show that the positive control which has PolyI:C but lacks the NS1 plasmid strongly drives the IFN-β promoter activation of the luciferase gene. Though the transfected cells showed equal expression of both NS1 proteins (Fig. 3b), importantly, the H11N1–NS1 influenced a greater level of inhibition (P < 0.05) of the IFN-β promoter activation compared to that of the H5N1–NS1 (Figs. 3a, S2). These results were further corroborated by immunoblotting the infected cell extracts with the anti-NS1 antibody which showed higher NS1 expression in H11N1-infected cells (Fig. 3c). The fact that the H11N1 virus-infected A549 cells produce greater amounts of NS1 protein relative to the H5N1 virus appears to contradict the replication kinetics shown in Fig. 1a. While it is acknowledged that greater protein expression levels do not necessarily result in higher infectious titers, differential regulation of translation of viral proteins for the LPAI and HPAI viruses might be a plausible explanation for the differences. It, however, is likely that the increased inhibition of IFN-β mRNA expression by H5N1 in comparison to H11N1 may be driven by an entity in synergy with NS1. The role of other gene segments is supported by an earlier study [4] and recent studies wherein viral polymerase proteins PB2, PA, and PB1-F2 affect virulence by inhibiting IFN-β expression [25,26,27,28,29]. A preliminary sequence comparison analysis of the above proteins of H5N1 and H11N1 showed differences in charged residues (Fig. 4a–c), suggestive of a possible role of H5N1 polymerases along with NS1 in modulating IFN signaling that needs to be addressed separately.

Ability of NS1 to inhibit IFN-β, expression of NS1 in infected cells, and pairwise alignment of NS1 proteins of H5N1 and H11N1 viruses. a Luciferase activities of A549 cells cotransfected with a luciferase reporter plasmid under the control of IFN-β promoter pGL4-IFN-β-luc and internal control (Renilla plasmid) followed by induction with PolyI:C. Data are presented as relative luciferase units ± SEM of three independent experiments. Asterisks indicate statistical significance by the Student’s t test (*P < 0.05; **P < 0.01). b Total cellular protein (25 µg) from the H11N1 and H5N1–NS1-transfected A549 cells and c cell lysate (equal protein) from A549 cells infected with H11N1 and H5N1 virus, were separated by SDS-PAGE and immunoblotted with anti-NS1 antibody. The graph in b represents fold expression of the transfected NS1 as quantified by densitometry analysis. The blot in c was stripped and re-probed with anti-β-actin antibody. The NS1 protein band signal intensity was normalized to the signal intensity of the respective β-actin and plotted as a graph. d NS1 protein sequences of H5N1 and H11N1 viruses were aligned and the different domains identified. Residues from 1–73 and 74–230 represent the RNA binding domain and the effector domain of the NS1 proteins, respectively. Residues highlighted in bold font and indicated with asterisk represent differences between H5N1 and H11N1–NS1. The 5 amino acid deletion in H5N1 NS1 is from residues 80–84. The CPSF30 binding sites at residues 103–106 and 144–188 have been shown by black horizontal lines


Pairwise alignment of PA, PB1, and PB2 proteins of H5N1 and H11N1 viruses. PA (a), PB1 (b), and PB2 (c) protein sequences of H5N1 and H11N1 viruses were aligned. Differences in charged residues between H5N1 and H11N1 are indicated by asterisk
Sequence and structure comparison of H5N1- and H11N1–NS1 proteins
Studies on the effect of different NS genes and single amino acid mutations from LPAI viruses on viral pathogenicity led to the identification of new residues in the NS gene important for mammalian pathogenicity [30]. Thus, in our study the significant differences observed in the ability of NS1 of the LPAI and HPAI viruses to inhibit IFN-β-prompted analysis of amino acid variations in NS1 and its structural comparison using bioinformatics tools. Results of pairwise alignment of H5N1 and H11N1 showed 13 amino acid variations and a 5 amino acid (80–84) deletion in H5N1 (Fig. 3d). The NS1 protein binds to the 30-kDa subunit of the cleavage and polyadenylation-specificity factor (CPSF30) to regulate cellular mRNA processing leading to general inhibition of the host antiviral response [16]. Notably, we found N171D (H5N1 vs H11N1) within the CPSF30 binding site (144–188) while M111V and T112A in close vicinity of another CPSF30 binding site (103–106), thus we propose that these changes might affect the binding of NS1 to CPSF30 in the H11N1 virus, thus modulating the host antiviral response. The role of the 5 amino acid residues (80–84) in IFN signaling is less plausible, as a study showed that deletion of these residues enhanced the virulence of H5N1 viruses [31], and these residues are not present in LPAI viruses, including the H11N1 virus in this study. In addition, the residues are not located within the CPSF30 binding sites of H11N1–NS1.
A 3D structure prediction of the NS1 proteins was obtained using the crystallographic structure of Influenza/A H6N6 NS1 (4OPH.PDB) as template using methods described earlier [23, 24]. The target template sequence identity was 93.33% for H5N1 and 98.70% for H11N1. The minimized energy of NS1 from H5N1 and H11N1 was − 9967.9 and − 10,103 kJ/mol, respectively. Superimposition of the 3D structures indicated a root-mean-squared deviation of 0.22 Å involving 760 backbone atoms, implying that the proteins have high structural similarity in terms of backbone fold. However, the above amino acid differences created local modifications in the surface contour and surface electrostatics (Fig. S3). The most prominent was N171D (H5N1 vs H11N1) which altered the local electrostatics around the CPSF30 binding site, wherein the H11N1–NS1 becomes more positively charged. The position of residue 171 in the backbone of the NS1 proteins is indicated in the ribbon mode display (Fig. S3). Since the specificity of protein–protein interaction depends on shape, complementarity of surface-contours of the participating proteins, surface electrostatics, and capacity to form H-bonds or chemical interaction, any change and resulting alteration of local surface contour and/or surface electrostatics affects protein–protein binding. Scrutiny of H11N1–NS1 sequences from GenBank and H5N1 strains (2006–2011) revealed N171 in the majority of H5N1 viruses and D171 in all H11N1 viruses. The presence of charged aspartic acid residue in H11N1 may modulate the binding affinity for CPSF30 to NS1. The inability of the NS1 protein of the 2009 pandemic influenza H1N1 virus to block host gene expression in human and swine cells has been attributed to reduced binding efficiency of NS1 to CPSF30 [32]. Hence, in our study, N171D and other differences between H5N1 and H11N1–NS1 and polymerase proteins may be responsible for the altered functional efficiency reflected by the differential response in inhibition of IFN-β promoter activation.
In conclusion, our study suggests that between the LPAI and HPAI viruses, the HPAI–H5N1 effectively attenuates IFN responses as a possible strategy to evade the host innate immune assault by a viral entity in synergy with NS1. However, which one among the IFN-antagonists (PB2, PA, or PB1) is responsible needs to be investigated. Though there are comparative studies between H5N1 and the human seasonal viruses H1N1 and H3N2 [25, 33], this study on LPAI and HPAI viruses adds significant data for mammalian pathogenicity studies on AI viruses. In the wake of emerging AI viruses and differential interferon responses to HPAI and LPAI viruses, the insight gained will be useful in designing further structural and protein interaction studies targeted at NS1 and polymerases for understanding host–virus interactions and molecular mechanisms governing differences in IFN responses.
References
P. Palese, M.L. Shaw, in Fields of Virology, ed. by D.M. Knipe, P.M. Howley (Lippincott Williams and Wilkins, Philadelphia, 2007), p. 1647
G. Neumann, Y. Kawaoka, Virology 479–480, 234–246 (2015)
D.L. Suarez, in Avian Influenza, 1st edn., ed. by D.E. Swayne (Blackwell, Iowa, 2008), p. 3
J.S. Peiris, M.D. de Jong, Y. Guan, Clin. Microbiol. Rev. 20, 243–267 (2007)
Z.M. Zhao, K.F. Shortridge, M. Garcia, Y. Guan, X.F. Wan, J. Gen. Virol. 89, 2182–2193 (2008)
R.G. Webster, W.J. Bean, O.T. Gorman, T.M. Chambers, Y. Kawaoka, Microbiol. Rev. 56, 152–179 (1992)
Y. Xu, A.M. Ramey, A.S. Bowman, T.J. DeLiberto, M.L. Killian, S. Krauss, J.M. Nolting, M.K. Torchetti, A.B. Reeves, R.J. Webby, D.E. Stallknecht, X.F. Wan, J. Virol. 91, e02208–e02216 (2017)
X. Xu, K. Subbarao, N.J. Cox, Y. Guo, Virology 261, 15–19 (1999)
M.L. Perdue, in Avian Influenza, ed. by D.E. Swayne (Blackwell, Iowa, 2008), p. 23
K. Ray, V.A. Potdar, S.S. Cherian, S.D. Pawar, S.M. Jadhav, S.R. Waregaonkar, A.A. Joshi, A.C. Mishra, Virus Genes 36, 345–353 (2008)
A.C. Mishra, S.S. Cherian, A.K. Chakrabarti, S.D. Pawar, S.M. Jadhav, B. Pal, S. Raut, S. Koratkar, S.S. Kode, Virol. J. 6, 26–34 (2009)
A.K. Chakrabarti, S.D. Pawar, S.S. Cherian, S.S. Koratkar, S.M. Jadhav, B. Pal, S. Raut, V. Thite, S.S. Kode, S.S. Keng, B.J. Payyapilly, J. Mullick, A.C. Mishra, PLoS ONE 4, e7846 (2009)
B. Opitz, A. Rejaibi, B. Dauber, J. Eckhard, M. Vinzing, B. Schmeck, S. Hippenstiel, N. Suttorp, T. Wolff, Cell. Microbiol. 9, 930–938 (2007)
G.A. Versteeg, A. Garcia, in Viruses and Interferon Current Research, ed. by K. Mossman (Caister Academic Press, Norfolk, 2011), p. 181
D. Jia, R. Rahbar, R.W.Y. Chan, S.M.Y. Lee, M.C.W. Chan, B.X. Wang, D.P. Baker, B. Sun, J.S. Malik Peiris, J.M. Nicholls, E.N. Fish, PLoS ONE 5, e13927 (2010)
B.G. Hale, R.E. Randall, J. Ortín, D. Jackson, J. Gen. Virol. 89, 2359–2376 (2008)
A. García-Sastre, A. Egorov, D. Matassov, S. Brandt, D.E. Levy, J.E. Durbin, P. Palese, T. Muster, Virology 252, 324–330 (1998)
S. Pawar, A. Chakrabarti, S. Cherian, S. Pande, M. Nanaware, S. Raut, B. Pal, S. Jadhav, S. Kode, S. Koratkar, V. Thite, A. Mishra, Virus Genes 41, 14–22 (2010)
S.S. Koratkar, S.D. Pawar, V.N. Shelke, S.D. Kale, A.C. Mishra, Indian J. Med. Res. 139, 782–785 (2014)
S. Mukherjee, S. Majumdar, V.C. Vipat, A.C. Mishra, A.K. Chakrabarti, Virol. J. 9, 149–160 (2012)
D.S. Tare, S.D. Pawar, J. Virol. Methods 224, 67–72 (2015)
K.J. Livak, T.D. Schmittgen, Methods 25, 402–408 (2001)
P. Shil, S.S. Chavan, S.S. Cherian, Bioinformation 7, 76–81 (2011)
P. Shil, S. Chavan, S. Cherian, Bioinformation 6, 266–270 (2011)
M. Matthaei, M. Budt, T. Wolff, PLoS ONE 8, e56659 (2013)
Z.T. Varga, A. Grant, B. Manicassamy, P. Palese, J. Virol. 86, 8359–8366 (2012)
M.J. Killip, E. Fodor, R.E. Randall, Virus Res. 209, 11–22 (2015)
J.C.D. Long, E. Fodor, J. Virol. 90, 8729–8738 (2016)
G. Kochs, A. Garcia-Sastre, L. Martinez-Sobrido, J. Virol. 81, 7011–7021 (2007)
I.-H. Kim, H.-J. Kwon, S.-H. Lee, D.-Y. Kim, J.-H. Kim, Vet. Microbiol. 175, 17–25 (2015)
J.X. Long, D.X. Peng, Y.L. Liu, Y.T. Wu, X.F. Liu, Virus Genes 36, 471–478 (2008)
B.G. Hale, J. Steel, R.A. Medina, B. Manicassamy, J. Ye, D. Hickman, R. Hai, M. Schmolke, A.C. Lowen, D.R. Perez, A. Garcia-Sastre, J. Virol. 84, 6909–6922 (2010)
H. Zeng, C. Goldsmith, P. Thawatsupha, M. Chittaganpitch, S. Waicharoen, S. Zaki, T.M. Tumpey, J.M. Katz, J. Virol. 81, 12439–12449 (2007)
D. Goubau, R. Romieu-Mourez, M. Solis, E. Hernandez, T. Mesplède, R. Lin, D. Leaman, J. Hiscott, Eur. J. Immunol. 39, 527–540 (2009)
D. Li, Z. Peng, H. Tang, P. Wei, X. Kong, D. Yan, F. Huang, Q. Li, X. Le, Q. Li, K. Xie, Clin. Cancer Res. 17, 3558–3568 (2011)
P.B. Devhare, S.N. Chatterjee, V.A. Arankalle, K.S. Lole, PLoS ONE 8, e63793 (2013)
CDC protocol of realtime RTPCR for influenza A viruses, http://www.who.int/csr/resources/publications/swineflu/realtimeptpcr/en/
Acknowledgements
We thank Ajit Rattan, Sachin Keng, Dinesh Singh, JPN Babu, SK Waghmare, AB Khare, and BSL3 staff for laboratory assistance, and P Aher, Drs. K Lole, P Yadav, and P Devhare for their help. We acknowledge the constant support of Dr. AC Mishra (Ex-Director) and Dr. DT Mourya, Director, NIV. We are grateful to Dr. Cecilia Dayaraj and Dr. Sarah Cherian for their critical comments. The work was supported by intramural funds from the Indian Council of Medical Research, New Delhi, India, and financial assistance to M. Thube from the Council of Scientific and Industrial Research, New Delhi, India and R. Kasbe from the University Grants Commission, New Delhi, India is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Conceived the idea and initiated the work: MMT & JM. Contributed to design of experiments: MMT, SDP, PS, & JM. Performed the experiments: MMT, RK, & PS (Bioinformatics). Contributed reagents/materials/analysis tools: JM, SDP, & PS. Analysis of experiments and results: MMT, PS, & JM. Contribution in generating graphics: MMT, RK, & AAP. Writing of the manuscript: MMT, PS, & JM. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This research work does not involve human participants and/or animals.
Informed consent
Not applicable as no human participants are involved.
Additional information
Edited by William Dundon.
This work was done in partial fulfillment of the Ph.D. thesis of M.M.T. submitted to the S. P. Pune University, Pune, India.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Thube, M.M., Shil, P., Kasbe, R. et al. Differences in Type I interferon response in human lung epithelial cells infected by highly pathogenic H5N1 and low pathogenic H11N1 avian influenza viruses. Virus Genes 54, 414–423 (2018). https://doi.org/10.1007/s11262-018-1556-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-018-1556-1