
Abstract
COVID-19 disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV2) has resulted in tremendous loss of lives across the world and is continuing to do so. Extensive work is under progress to develop inhibitors which can prevent the disease by arresting the virus in its life cycle. One such way is by targeting the main protease of the virus which is crucial for the cleavage and conversion of polyproteins into functional units of polypeptides. In this endeavor, our effort was to identify hit molecule inhibitors for SARS-CoV2 main protease using fragment-based drug discovery (FBDD), based on the available crystal structure of chromene-based inhibitor (PDB_ID: 6M2N). The designed molecules were validated by molecular docking and molecular dynamics simulations. The stability of the complexes was further assessed by calculating their binding free energies, normal mode analysis, mechanical stiffness, and principal component analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronaviruses comprise a large family of zoonotic viruses that have potential to infect humans via animal intermediaries [1]. Coronaviruses pose a continuous threat to the mankind because of their ability to emerge unpredictably and periodically, thereby spreading rapidly and inducing serious infectious pandemic disease. The world previously witnessed outbreaks of coronavirus disease caused by other similar species, namely SARS-CoV in 2002 and 2003 in China [2]; MERS-CoV in 2012 in the Middle East [3]; HCoV-OC43 [4, 5] in 1967 in Salisbury, UK; HCoV-229E [6] in 1966 in African bats; HCoV-HKU1 [7] in 2005 in Hong Kong; and HCoV-NL63 [8, 9] in 2002–2003 across Asia, Europe, and North America. During the last 2 years, the novel human coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) that causes coronavirus disease 2019 (COVID-19) resulting in an acute infection in the respiratory system [1], has become a global pandemic. All these viruses belong to the subfamily Coronavirinae in a family Coronaviridae. These are nonsegmented, enveloped, positive‐sense single‐stranded RNA virus genomes [10], and the human coronaviruses have the genomic size ranging from 29 to 31 kilobases. The RNA genome of the human SARS-CoV2 is 82% identical to that of human SARS-CoV [11] and shares considerable similarity in the genome; hence, it has been named as SARS-CoV2 [12]. The SARS-CoV2 genome encodes open-reading frames (ORFs) ORF1a and ORF1ab that encode for polyproteins. These polyproteins are further processed into 16 nonstructural proteins (NSPs), NSP1 to NSP16, which constitute the replicase-transcriptase complex. Various enzymes including the main protease also called as 3-chymotrypsin-like protease (NSP5), papain-like protease (NSP3), and RNA-dependent RNA polymerase (NSP12) are present in this complex among other proteins [13]. The virion contains a nucleocapsid made up of genomic RNA and phosphorylated nucleocapsid protein, buried inside the phospholipid bilayers and surrounded by spike protein on the membrane of virus. A type-3 transmembrane glycoprotein also known as membrane protein and the envelope protein are situated among the spike proteins in the virus envelope [10].
The most prominent symptom reported in SARS-CoV2 is respiratory distress with some patients requiring intensive care, high-flow oxygen therapy, and invasive and noninvasive ventilation. The condition could worsen due to respiratory failure leading to the death of the patient [14]. It was also observed that patients with comorbidity pose a greater risk of developing severe infection which may also result in death of an individual [15]. More than 530 million COVID-19 cases and close to 6.29 million deaths have been reported during the last 2 years and 5 months. Despite the availability of vaccines and large populations in various countries being already vaccinated, the re-emergence of SARS-CoV2 in the mutated forms as witnessed in the case of beta [16] and omicron [17] variants poses a continuous threat of COVID-19 disease. There have been continuous research efforts to develop antiviral agents against members of Coronaviridae family which suggested that the human receptor, angiotensin-converting enzyme 2 that facilitates entry of virus into the host, RNA-dependent RNA polymerase and main protease, helicase, and papain-like protease, may be represented as suitable drug targets [11, 18,19,20,21,22,23].
Considering the functional significance of the main protease, i.e., to carry out processing of polyproteins into NSPs, in the viral life cycle, and combined with the absence of closely related homologues in humans, main protease serves as an attractive target for the design of antiviral drugs. Inhibiting the viral activity of this enzyme would block the replicase-transcriptase complex and therefore disrupt the viral life cycle. These targets are a choice for inhibition purpose also in SARS-CoV and MERS-CoV, and hence, constant work has been carried out either by computational methods or experimental investigations to find novel molecules which can interfere with virus life cycle by inhibiting the main protease. For example, glycosylated flavonoids display a strong inhibitory activity on SARS-CoV2 main protease [24]. Some natural polyphenolic compounds, i.e., glucogallin, mangiferin, and phlorizin, may have the ability to be used as safe protease inhibitors, which may act by inhibiting type-2 transmembrane serine protease and main protease [25]. In silico approaches have identified three FDA-approved drugs, namely remdesivir, saquinavir, darunavir, and two small molecules such as flavone and coumarin derivatives that act as potential inhibitors of human SARS-CoV2 main protease [26]. Few drugs were identified and tested for treatment of COVID-19 disease such as chloroquine, lopinavir, nafamostat, hydroxychloroquine, ritonavir, camostat, corticosteroids, and sarilumab [27]. Also a combination of drugs such as noscapine and hydroxychloroquine demonstrated a strong binding affinity towards main protease [28]. Numerous drug candidates such as ebselen, disulfiram, carmofur, α-ketoamides, and peptidomimetic aldehydes 11a/11b have been discovered which inhibit the main protease activity by providing interference with the maturation process of NSPs [11, 22, 29, 30]. Computational studies have identified some of the NCI natural products as SARS-CoV2 main protease inhibitors [31].
The present work focuses on finding suitable inhibitors for the main protease in SARS-CoV2 using the computational methodologies in fragment-based drug discovery (FBDD). The FBDD methodology initially employs virtual screening of chemical fragments with low molecular weight and less chemical complexity and targets the sub-pockets within the receptor binding site [32]. The selection of fragments in a library generally follows “rule of three” criterion, i.e., molecular weight \(\le\) 300, hydrogen bond donor and acceptor \(\le\) 3, and lipophilicity index (CLogP) \(\le\) 3 [33, 34]. Furthermore, it is desirable that the number of rotatable bonds is ≤ 3, and the polar surface area (PSA) is ≤ 60 [35]. Such fragment hits are considered to be more suitable before the “hit-to-lead optimization” since they have reduced complexity, thereby allowing more freedom for optimization of various properties of the fragment hits in a multidimensional manner [32]. Some attractive features like favorable physical, pharmacokinetics, and toxicity properties can be integrated in the development of fragments [36]. These fragments function as building blocks, combined into compounds that typically recognize the protein binding site. The process of optimization of fragments can be done by linking the two independent fragments together or by adding functional groups in each step to make improvements to the molecules [32]. These compounds are further elaborated such that they bind the receptor via several nonbonding interactions. Methodologies in FBDD have rapidly led to the identification of several FDA-approved drugs such as INQOVI, erdafitinib [37], ribociclib, vemurafenib [38], pexidartinib [39], and venetoclax [40,41,42].
The inhibitor 3WL complexed with SARS-CoV2 main protease was used as the reference structure on which FDA-approved drug fragments were employed in order to obtain new lead molecules that could bind the active site of the receptor. Molecular docking, molecular dynamics (MD) simulations, and trajectory analyses were employed in order to propose new hit molecules to bind the SARS-CoV2 main protease.
Materials and methods
Protein preparation
The crystal structure of SARS-CoV2 main protease was solved at 2.2 Å resolution, and its three-dimensional structure complexed with inhibitor is deposited in the protein structure databank (PDB) with the PDB_ID: 6M2N [43]. The crystal structure of the main protease is a homodimer; chain A was selected for the computational studies as it did not have any missing residues. Crystal waters were deleted, hydrogen atoms were added to complete the valency, and geometry optimization of the structure was done using minimize structure tool in the UCSF Chimera [44] version 1.12 for further studies.
Inhibitor design
We used Auto Core Fragment in silico Screening (ACFIS) 2.0 web server available at http://chemyang.ccnu.edu.cn/ccb/server/ACFIS/ [45] that comprises FDA drug fragment database with 2883 fragment molecules for FBDD [45, 46]. This server works on the principle of pharmacophore-linked fragment virtual screening approach. ACFIS has three computational modules. PARA_GEN is a tool to generate parameters for ACFIS using the AMBER tools; CORE_GEN tool considers the protein–ligand complex and proceeds to deconstruct the ligand structure into fragments by cleavage at single bond based on retrosynthetic analysis by using decomposition and identification of molecules program [47] to derive core fragment structure from a bioactive ligand. CAND_GEN is a tool to link fragments to the core fragment structure and generate candidate molecules. The fragments in the library will be linked to the junction of the core fragment structure at the position of cleavage. ACFIS server creates new molecules based on growing algorithm by determining the core fragment structure followed by automatic linking of each of the fragments for improving the binding affinity. Finally, the molecules are ranked based on their binding affinities. Output structure files and related data were downloaded. These molecules thus identified were used for further molecular docking studies.
Molecular docking
Molecular docking is a tool to predict molecular recognition between a small molecule and a target macromolecule in terms of binding modes and binding affinity. Molecular docking involves generating the most favorable binding mode of a ligand to its target macromolecule. Virtual screening of molecules by molecular docking was performed by using PyRx [48] and AutoDock 4.2 [49] software. Both the docking methods were validated by docking 3WL into the active site of main protease 6M2N as seen in Supplementary Fig. 1A, B. The interacting residues within a radius of 5 Å with respect to reference molecule 3WL were considered for generation of grid box. The protein macromolecule and ligands were imported using input molecule tools in PyRx and were converted from .pdb into .pdbqt format. Grid box was generated and adjusted so that it includes the active site residues at the binding site, with the exhaustiveness value of 10. The results were analyzed for determining the best conformers with lowest binding energy (kcal/mol) and hydrogen bond interactions with the active site residues. The result output was exported to a file for visualization and analysis purpose using discovery studio visualizer.
The best ranked molecules were subjected to another round of docking studies using AutoDock 4.2 tools in order to dock them inside a 5 Å cavity defined around the reference molecule 3WL. The protein was cleaned by deletion of heteroatoms and water molecules, torsions were set, polar hydrogens were added, and structure was saved in .pdbqt format. Similarly, the designed molecules were loaded, and torsions were set and saved in .pdbqt format. The calculations for protein–ligand flexible docking were performed using the Lamarckian genetic algorithm. The maximum number of generations simulated during each GA run was set to 27,000, and the rates of the gene mutation and crossover were set to 0.02 and 0.8, respectively. The threshold RMSD value was set to 2 Å, and the number of docking poses generated for each molecule was set to 10 for AutoDock as well as PyRx. A grid box with the dimensions of X: −32.464 Å, Y: −62.314 Å, and Z: 43.532 Å and a grid spacing of 0.375 Å was used with exhaustiveness value of 10. The conformers obtained were ranked based on the nature of interactions, root-mean-square deviation (RMSD) values, and binding affinity scores.
Drug likeness
The absorption, distribution, metabolism, and excretion (ADME) parameters describe the pharmacokinetic properties associated with the ligand molecules and have a crucial role to play in the process of drug discovery and development. The ADME properties of the docked molecules were measured using the SwissADME server [50]. SwissADME server is a free webtool to predict the drug-likeness properties of small molecules; it displays the pharmacokinetic properties in the form of topological polar surface area (TPSA), consensus LogPO/W, Log S, and Log KP, and synthetic accessibility values.
Molecular dynamics simulations
Proteins are inherently flexible biomolecules which combine with a ligand to form the protein–ligand complex; this complex attains stability mediated by various nonbonding interactions such as van der Waals (vdW), electrostatic (el), hydrogen bonding, and hydrophobic. These kinds of interactions can be studied using MD simulations. Proteins identified by X-ray diffraction consider static conditions, while the MD simulations can be used to find the consistency in non-covalent interactions and the stability of protein–ligand complexes. The MD simulation studies were run for the apo form of main protease as well as with the reference molecule bound in the complex form using GROMACS 5.1.4 [51] software for 250 ns. The force fields for protein were generated using AMBER ff99SB [52], and force fields for hit molecules and reference ligand were generated in antechamber [53] using ACPYPE [54] scripts. The complexes were immersed in a cubic box filled with water molecules as simple point charge (SPC) [55] model. The neutralization of the system was done by adding Na+ and Cl− ions. This was followed by energy minimization using steepest descent method for 50,000 steps to relieve the system from short-range bad contacts. The equilibration of the system is performed in two steps by allowing the system to get equilibrated until it reaches 300-K temperature in the constant number, volume and temperature (NVT) step and also until it attains the suitable density in constant number, pressure and temperature (NPT) (1 atm, 300 K) step. The NVT and NPT equilibration were performed for 100 ps timescale to energy minimize the system. For temperature and pressure couplings, V-rescale thermostat [56] and Parrinello and Rahman methods [57] were used respectively. The long-range electrostatics were handled using the particle mesh Ewald method [58]. The Lennard–Jones interactions, real space interactions, were truncated at 9 Å, and hydrogen bonds were constrained using LINCS algorithm [59]. The final production run was performed for 250 ns timescale involving 125 million steps. The final models for all molecular systems were obtained by averaging the snapshots generated from the trajectories of MD simulations. The RMSD and root-mean-square fluctuations (RMSF) were calculated using gmx rms and gmx rmsf commands, respectively, of GROMACS to study the conformational variations in the protein bound to reference and hit molecule complexes.
Binding free energies
The interaction energy between the protein and ligand is vital to study about the overall stability of the complex and to assess how well the ligand fits into the protein binding site. Also, the contribution from amino acid residues which form the ligand binding site in complex formation can be estimated from binding free energy. The Poisson-Boltzmann or Generalized Born and Surface Area Continuum Solvation (MM-PBSA and MM-GBSA) [60] are methods that can estimate the free energy of binding of small molecules to the protein. The output trajectories from GROMACS with the g_mmpbsa tool were used to calculate binding free energies for all the complexes. The linear interaction energy (LIE) method [61], which is based upon estimations of force fields in the receptor-ligand interactions and thermal conformational sampling, was used for calculation of ligand binding free energies. The gmx lie tool was utilized to compute free energy estimate based on analysis from nonbonded energies.
Generally, the binding free energy of a protein in complex with a ligand is expressed as [62, 63]:
Here, Gcomplex is the total free energy of protein–ligand complex, and Gprotein and Gligand are the total free energies of isolated forms of protein and ligand in the solvent, respectively.
In the LIE approach, the binding free energy is estimated according to the following equation and is divided into polar and nonpolar contributions [64].
where ⟨⟩ depicts the MD averages for the nonbonded vdW and el interactions of the ligand with its surrounding environment (l-s). The Δ's depict change in the MD averages when ligand is transferred from solution (free state) to the binding site of the solvated receptor (bound state). The coefficients α and β corresponding to parameters for nonpolar and polar interactions, respectively, are scaling factors for these energies, and γ is a constant term which could also be implemented as surface-area dependent [65, 66]. The values taken for α, β, and γ were 0.181, 0.5, and 0, respectively.
Normal mode analysis and mechanical stiffness
Protein dynamics is very essential to understand molecular events in a cell such as ligand recognition, binding, and transport [67]. The ensembles of structures can provide information about the structural variations important for biological activity [68, 69]. We used software suite of programs in ProDy [67] to develop the elastic network model-based normal mode analysis (NMA), and dihedral angles were taken as independent variables for all molecular systems. The mechanical stiffness plots were developed for all molecular systems using anisotropic network model [70].
Principal component analysis
Principal component analysis (PCA) is a covariance-matrix-based mathematical technique to find global and correlated motions in atomic simulations of protein [71]. The overall motion of main protease was assessed through PCA technique by generating a 3N × 3N covariance matrix followed by its diagonalization to construct eigenvectors. The technique was employed for all the simulated molecular systems using MODE-TASK [72], and calculations were carried out from 0 to 250 ns MD simulations trajectories.
Results and discussions
Overall fold and active site of SARS-CoV2 main protease
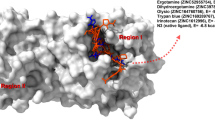
Several crystal structures of SARS-CoV2 main protease in complex with peptide-based inhibitors are available in the Protein Data Bank (PDB) with IDs as follows: 6XMK, 6XBH, 7BRP, 7CB7, and 7CBT. We have selected the crystal structure of SARS-CoV2 main protease in complex with a small molecule inhibitor (3WL), having PDB_ID: 6M2N [43] as the reference system. The three-dimensional structure of SARS-CoV2 main protease contains three domains. The domain 1 (8–101 amino acid residues) and domain 2 (102–184) fold into β-barrels and bear close resemblance to the chymotrypsin, whereas domain 3 (201–306) is mainly composed of α-helices. The substrate binding region, which is located at the cleft of domains 1 and 2, comprises of catalytic dyad His41 and Cys145, in which Cys serves as a nucleophile, while His acts as a proton acceptor. There are two deeply buried subsites (S1 and S2) and three shallow subsites (S3–S5) in addition to the catalytic site that also contribute to the active site of main protease. The S1 subsite is composed of Phe140, Gly143, Cys145, His163, Glu166, and His172, and S2 subsite is composed of Thr25, His41, and Cys145 amino acid residues. The shallow subsites S3–S5 are formed by His41, Met49, Met165, Glu166, and Gln189 amino acid residues [26]. This molecule makes multiple hydrogen bonds with side chain and main chain atoms in the protein active site. The small molecule 3WL chromene moiety forms hydrogen bonding interactions with residues Leu141, Gly143, Ser144, and Glu166. The phenyl ring moiety is embedded in a hydrophobic pocket formed by His41, Cys44, Met49, and Tyr54 as shown in Fig. 1.

The nonbonding interactions of 3WL in the binding site of SARS-CoV2 main protease (PDB_ID: 6M2N)
Inhibitor design and molecular docking
Here, we used SARS-CoV2 main protease complexed with 3WL molecule. The molecule 3WL was divided into two fragments (as shown in Fig. 2): fragment 1 (chromene ring) and fragment 2 (phenyl ring) that have binding affinity values −15.73 kcal/mol and −4.95 kcal/mol and ligand efficiency values (LE) 1.12 (fragment 1) and 0.82 (fragment 2) as shown in Supplementary Table 1 using ACFIS server. The molecular size can be eliminated by using LE rather than potency. The LE of fragment 1 (1.12) is relatively higher than the fragment 2 (0.82). However, comprehensively considered with Gbind, LE, and new fragment growing spaces, fragment 2 was selected as a core fragment to perform CAND_GEN calculation. In this molecule, also, since the 5,6,7-trihydroxy-2-phenyl-4H-chromen-4-one chromene ring makes several hydrogen bonding interactions with the protein active site, only the phenyl ring was searched for FBDD. The screening and linking was performed at fragment 2 (phenyl) ring position. The screening of FDA-approved fragment library with 2883 fragment molecules with integrated energy calculations identified 29 fragments. These fragments were successfully joined with the chromene ring in fragment 1 to form 29 new molecules. The output coordinates were downloaded and used for docking studies. The 29 hit molecules obtained by FBDD approach using ACFIS server were docked into the active site of main protease using PyRx and AutoDock to study their binding interactions that are shown in Table 1 and Supplementary Table 2. Also, the cluster RMSD mostly ranged from 0.00 to 1.76 Å, wherein the lowest energy docking pose from the largest cluster was selected. In other terms, the best conformer from the possible generated conformers was selected based on RMSD value and the binding affinity score. The ten best molecules selected from both the docking methods show binding affinity in the range −9.0 to −6.8 kcal/mol (AutoDock) and −8.5 to −6.7 kcal/mol (PyRx), while the reference molecule 3WL had docking score of −7.7 and −7.2 kcal/mol using AutoDock and PyRx, respectively, as shown in Table 1. The intermolecular interactions such as hydrogen bonding, vdW, el, and hydrophobic observed due to the docked hit molecules within the active site residues such as His41, Leu141, Asn142, Gly143, Ser144, Cys145, and Glu166 also favored the selection of best conformers. The best ten molecules from the docking studies were used for drug-likeness studies.

The fragment-based approach on 3WL for selection of hit molecules. A 3WL B Fragment 1 (chromene ring) C Fragment 2 (phenyl ring)
Drug-likeness properties
The ADME properties of the selected 10 molecules and reference molecule are tabulated in Table 2. The synthetic accessibility values for hit molecules lie within 2.92–4.03 which is indicative of their ease of synthesis. The topological surface area lies between 90.90 and 120 Å2, and the lipophilicity parameter consensus LogP is less than 3. Log S values for the hit molecules are comparable with the reference molecule which indicates that their water solubility is similar with the reference molecule. The skin permeation of hit molecules expressed as Log Kpvalue is also in good agreement with respect to reference molecule. All the qualified molecules were used for further stability studies by MD simulations.
Molecular dynamics simulations
The main protease 6M2N complexed with reference molecule 3WL and hit molecules were subjected to 250 ns of MD simulations using GROMACS 5.1.4. Out of the ten molecules studied, four molecules (Hit I, Hit II, Hit III, Hit IV with 1H-benzo[d]imidazol-5-yl, 3H-indol-3-yl, isoxazol-5-ylmethyl, 6-fluoro-1,2,3,4-tetrahydronaphthalen-2-yl formate groups, respectively) exhibit stability throughout 250 ns MD simulations. The structural changes of these four molecular systems were analyzed by comparison of initial and average structures. In this manuscript, we report the screened molecules designed from FBDD that show good binding affinity and nonbonding interactions with SARS-CoV2 main protease. The results demonstrated that protein attains stability when it binds with screened-in hit molecules and maintains the hydrogen bonding interactions with important amino acids compared with reference molecule (3WL). The RMSD plots in Fig. 3a revealed that the structures attained stability after 10 ns of MD simulations. The main protease when complexed with Hit I, Hit II, Hit III, and Hit IV indicated greater structural stability of these four complexes. The hit molecules (I, III, IV) exhibit lower RMSD (lower than 0.25 nm), whereas Hit II exhibited RMSD value of 0.27 nm, and the reference molecule 3WL showed relatively higher RMSD (~ 0.28 nm) as shown in Fig. 3a. The RMSD of apo protein is higher (~ 4 Å) when compared with the complexes of reference and the hit molecules identified in this work, suggesting that binding of both reference and hit molecules enhances the structural stability of SARS-CoV2 main protease. In the ligand RMSD plots (Fig. 3b), we observed an abrupt change in the RMSD of Hit IV. From the examination of Hit IV-active site interacting residues, we observed that around 160–170 ns of MD simulations Glu166, His163, and Ser144 form hydrogen bonding interactions, whereas during 170–190 ns, Met49, Leu141, Glu166, and His172 form hydrogen bonding interactions. The conformational changes in Hit IV and therefore altered interacting amino acid residues in the binding pocket of SARS-CoV2 main protease may be responsible for abrupt change in its RMSD value.

a The RMSD plot of SARS-CoV2 main protease apo and when complexed with reference and identified hit molecules (I to IV). b The RMSD plots of ligand, reference molecule, 3WL, and four hit molecules (I to IV) when complexed with SARS-CoV2 main protease
The RMSF plots analyzed the residual fluctuations of protein during MD simulations. Higher fluctuations are observed in the regions: Cys44-Asn53, Phe140-Ser144, Ile152-Val157, and Gln273-Thr280 that are away from the active site and dimer interface of the SARS-CoV2 main protease. The RMSF plot of proteins is given in Fig. 4. It can be seen from the RMSF plot that most regions of main protease have fluctuations below 2 Å. The amino acid region from Cys44 to Asn53 that forms a loop-helix in domain 1 has high fluctuations in the apo protein (up to 4.5 Å), while it is stabilized in the presence of reference and hit molecules (RMSF below 2.5 Å). The lower fluctuations could be attributed to the stability gained by the SARS-CoV2 main protease reference and hit molecule complexes resulting from the close proximity and the presence of nonbonding interactions in this region (Supplementary Fig. 2). It is likely that this loop-helix is functioning as a flap in order to open and close the active site of SARS-CoV2 main protease. It is intriguing to observe that both the apo and SARS-CoV2-ligand complexes have a slightly higher RMSF up to 2.6 Å in domain 3 (Gln273-Thr280) that is away from the ligand binding site. The higher flexibility in domain 3 was also observed in our previous studies [31]. The stability of the identified hit molecules is comparable to the reference inhibitor, 3WL. These analyses showed that the docking poses of the hit molecules are accurate and comparable with the reference inhibitor molecule. The intermolecular hydrogen bonding interactions were stabilized throughout the 250 ns MD simulations as shown in Supplementary Fig. 3. The average structure taken from 0 to 250 ns MD simulations trajectories were analyzed for intermolecular hydrogen bonding interactions for reference and hit molecules and shown in Table 3. The occupancies of the interactions throughout the MD simulations and in the last 20 ns are provided in the Supplementary Table 3. During the last 20 ns of MD simulations, the reference 3WL, Hit I, Hit II, and Hit IV retain stable hydrogen bonding interactions with SARS-CoV2 main protease, while the hydrogen bonding interactions are less stable when complexed with Hit III.

The RMSF plots of apo and complexes of SARS-CoV2 main protease with reference and hit molecules (I to IV)
Normal mode and mechanical stiffness analysis
The NMA provides essential information about the ensembles of structures attained by protein during MD simulations run and hence illustrates the overall protein dynamics. The structural changes of protein also determine the pattern of mobility across its various domains and regions. From the RMSF plot, the regions in protein structure showing greater fluctuations such as Cys44-Asn53, Phe140-Ser144, Ile152-Val157, and Gln273-Thr280 were observed. The same observation was also demonstrated through the NMA and mechanical stiffness plots in all molecular systems. We obtained ten normal modes for each molecular system from MD trajectories, selected first three modes, and made comparisons with the apo structure of main protease. The regions from Cys44-Asn53, Phe140-Ser144, Ile152-Val157, and Gln273-Thr280 displayed higher mobility in apo protein, and that was slightly decreased in protein complexes with hit molecules (Fig. 5). Therefore, from this method, we studied about the conformational changes in apo and hit molecules bound complexes and located the flexible regions in the structure of SARS-CoV2 main protease.




NMA and mechanical stiffness plots of SARS-Cov2 main protease. A Apo. B 3WL. C Hit I. D Hit II. E Hit III. F Hit IV
The mechanical stiffness was analyzed for all the molecular systems, and the structural deviations were compared with the apo form of the main protease (Fig. 5). The flexibility caused by residues involving Cys44-Asn53, Phe140-Ser144, Ile152-Val157, and Gln273-Thr280 displayed lower effective stiffness for all molecular systems. This can be more clearly understood from the mean plots of mechanical stiffness which showed effective spring constant value of 8 k (a.u) for fluctuating regions and value greater than 12 k (a.u) for the stable regions in the protein structure. The mean plots also indicate that protein-bound hit complexes had increased stability than the apo form that is reflected by mean values greater than 12 k (a.u) and less than 8 k (a.u). These values suggest that protein shows elastic behavior due to the fluctuating residues in all complexed molecular systems studied. Therefore, from NMA and mechanical stiffness plots, we observed mechanically weak behavior caused by fluctuating regions from Cys44-Asn53, Phe140-Ser144, Ile152-Val157, and Gln273-Thr280.
Principal component analysis
The conformational changes in the apo form, reference 3WL, and hit molecules bound molecular systems were monitored during the entire simulations run of MD trajectories through PCA scatter plots. The PCA scatter plots for all the molecular systems are depicted in Fig. 6. From the plots, it is observed that the molecular system such as 3WL and Hit III bound complex showed large protein conformational changes and therefore vary distribution in scatter plots.

PCA scatter plots for SARS-CoV2 main protease. A Apo. B 3WL. C Hit I. D Hit II. E Hit III. F Hit IV
PCA calculations reveal the conformational changes in a protein as a function of time from the MD simulations trajectories. PCA was performed on 250 ns MD simulations trajectories to understand the alterations in the SARS-CoV2 main protease structure. We generated 25,000 frames for each molecular system, and the motion of Cα atoms in each molecular system was monitored. The conformational ensembles of the main protease in all molecular systems under study were analyzed by projecting the trajectories of PC1 and PC2 into a two-dimensional space. When these are mapped onto each other, the structures with a high degree of similarity cluster together. The conformational changes of the SARS-CoV2 main protease in apo form, 3WL, and hit molecules bound molecular systems were monitored. We observed that the first two principal components (PC1 and PC2) capture majority of the variance in the original distribution of conformational ensembles in the molecular systems as shown in Fig. 6. The distribution of Cα atoms was greater for 3WL and Hit III bound molecular system which indicates that greater conformational changes of protein are observed, and also, apo protein shows significant distribution throughout MD simulations. This demonstrated that the conformational distributions of main protease bound with 3WL and Hit III were remarkably different from other molecular systems. The frequencies of PCA scatter plots were quantified, and the highest frequency is observed in 3WL and Hit III bound main protease, indicative of their higher protein conformational changes compared to other molecular systems.
Binding free energies
The binding free energy, vdW, el, polar, and apolar solvation energies were calculated and compared to the reference inhibitor molecule in order to rank the hit molecules as high- or low-potent inhibitors. The binding free energies of all the complexes are depicted in Table 4. On the whole, from the binding free energy studies, we infer that the designed hit molecules using FBDD are comparable to the reference molecule, 3WL, in particular Hit III is closer to the reference molecule.
Conclusions
The crystal structure of SARS-CoV2 main protease (PDB_ID: 6M2N) in complex with inhibitor molecule 3WL was selected as the reference structure to design new hit molecules based on FBDD approach. The FBDD approach identified 29 hit molecules which retained fragment 1 of 3WL and fragment 2 replaced by different chemical entities. These molecules were studied for their binding affinity in the active site by molecular docking as well as for drug-likeness properties, and the best four hit molecules were selected containing fragment 2 as 1H-benzo[d]imidazol-5-yl, 3H-indol-3-yl, isoxazol-5-ylmethyl, and 6-fluoro-1,2,3,4-tetrahydronaphthalen-2-yl formate groups. MD simulations and binding free energy calculations proposed the four hit molecules to be stable in complexed form and fit well inside the active site of SARS-CoV2 main protease.
Availability of data and material
All data is included in the manuscript and provided as Supplementary information.
Code availability
Not applicable.
References
Carlos WG, Dela Cruz CS, Cao B, Pasnick S, Jamil S (2020) Novel Wuhan (2019-nCoV) coronavirus. Am J Respir Crit Care Med P7–P8
Drosten C, Günther S, Preiser W, Van Der Werf S, Brodt HR, Becker S, Doerr HW (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348(20):1967–1976
Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367(19):1814–1820
Kaye HS, Marsh HB, Dowdle WR (1971) Seroepidemiologic survey of coronavirus (strain OC 43) related infections in a children’s population. Am J Epidemiol 94(1):43–49
Hendley JO, Fishburne HB, Gwaltney JM Jr (1972) Coronavirus infections in working adults: eight-year study with 229 E and OC 43. Am Rev Respir Dis 105(5):805–811
McIntosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM (1967) Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc Natl Acad Sci USA 57(4):933
Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Yuen KY (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79(2):884–895
Van Der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Berkhout B (2004) Identification of a new human coronavirus. Nat Med 10(4):368–373
Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, De Jong JC, Simon JH, Osterhaus AD (2004) A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci 101(16):6212–6216
Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, Zhang Q (2020) Coronavirus infections and immune responses. J Med Virol 92(4):424–432
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Hilgenfeld R (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368(6489):409–412
Gorbalenya AE, Baker SC, Baric R, Groot RJD, Drosten C, Gulyaeva AA, Ziebuhr J (2020) Severe acute respiratory syndrome-related coronavirus: the species and its viruses–a statement of the Coronavirus Study Group
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, Tummino TA (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 1–13
Li YC, Bai WZ, Hashikawa T (2020) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol 92(6):552–555
Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, Zhou Y (2020) Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis 94:91–95
Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira IA, Gupta RK (2021) SARS-CoV-2 B. 1.617. 2 Delta variant replication and immune evasion. Nature 599(7883):114–119
Karim SSA, Karim QA (2021) Omicron SARS-CoV-2 variant: a new chapter in the COVID-19 pandemic. Lancet 398(10317):2126–2128
Ton AT, Gentile F, Hsing M, Ban F, Cherkasov A (2020) Rapid identification of potential inhibitors of SARS‐CoV‐2 main protease by deep docking of 1.3 billion compounds. Mol Inf
Borgio JF, Alsuwat HS, Al Otaibi WM, Ibrahim AM, Almandil NB, Al Asoom LI, AbdulAzeez S (2020) State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch Med Sci 16(3):508
Beck BR, Shin B, Choi Y, Park S, Kang K (2020) Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J
Ullrich S, Nitsche C (2020) The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett 127377
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Yang H (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293
Nimgampalle M, Devanathan V, Saxena A (2020) Screening of chloroquine, hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. J Biomol Struct Dyn 1–13
Cherrak SA, Merzouk H, Mokhtari-Soulimane N (2020) Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PLoS ONE 15(10):e0240653
Singh R, Gautam A, Chandel S, Ghosh A, Dey D, Roy S, Ghosh D (2020) Protease inhibitory effect of natural polyphenolic compounds on SARS-CoV-2: an in silico study. Molecules 25(20):4604
Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z (2020) Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dyn 1–10
Shaffer L (2020) 15 drugs being tested to treat COVID-19 and how they would work. Nat Med
Kumar N, Awasthi A, Kumari A, Sood D, Jain P, Singh T, Chandra R (2022) Antitussive noscapine and antiviral drug conjugates as arsenal against COVID-19: a comprehensive chemoinformatics analysis. J Biomol Struct Dyn 40(1):101–116
Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, Liu H (2020) Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368(6497):1331–1335
Jin Z, Zhao Y, Sun Y, Zhang B, Wang H, Wu Y, Rao Z (2020) Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol 27(6):529–532
Durgam L, Guruprasad L (2022) Computational studies on the design of NCI natural products as inhibitors to SARS-CoV-2 main protease. J Biomol Struct Dyn (Accepted)
Kumar A, Voet A, Zhang KYJ (2012) Fragment based drug design: from experimental to computational approaches. Curr Med Chem 19(30):5128–5147
Congreve M, Carr R, Murray C, Jhoti H (2003) A‘rule of three’for fragment-based lead discovery? Drug Discovery Today 8(19):876–877
Jhoti H, Williams G, Rees DC, Murray CW (2013) The‘rule of three’for fragment-based drug discovery: where are we now? Nat Rev Drug Discovery 12(8):644–644
Brown N (2015) In silico medicinal chemistry: computational methods to support drug design. Royal Society of Chemistry
de Souza Neto LR, Moreira-Filho JT, Neves BJ, Maidana RLBR, Guimarães ACR, Furnham N, Silva FP Jr (2020) In silico strategies to support fragment-to-lead optimization in drug discovery. Front Chem 8:93
Murray CW, Newell DR, Angibaud P (2019) A successful collaboration between academia, biotech and pharma led to discovery of erdafitinib, a selective FGFR inhibitor recently approved by the FDA. MedChemComm 10(9):1509–1511
Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P (2012) Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discovery 11(11):873–886
Lamb YN (2019) Pexidartinib: first approval. Drugs 79(16):1805–1812
Erlanson DA, de Esch IJ, Jahnke W, Johnson CN, Mortenson PN (2020) Fragment-to-lead medicinal chemistry publications in 2018. J Med Chem 63(9):4430–4444
Baker M (2013) Fragment-based lead discovery grows up: with multiple drug candidates in the clinic that originated from fragment-based lead discovery, the approach of starting small has become big. Nat Rev Drug Discovery 12(1):5–8
Denis JDS, Hall RJ, Murray CW, Heightman TD, Rees DC (2021) Fragment-based drug discovery: opportunities for organic synthesis. RSC Medicinal Chemistry 12(3):321–329
Su HX, Yao S, Zhao WF, Li MJ, Liu J, Shang WJ, Xu YC (2020) Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin 41(9):1167–1177
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Hao GF, Jiang W, Ye YN, Wu FX, Zhu XL, Guo FB, Yang GF (2016) ACFIS: a web server for fragment-based drug discovery. Nucleic Acids Res 44(W1):W550–W556
Yang JF, Wang F, Jiang W, Zhou GY, Li CZ, Zhu XL, Yang GF (2018) PADFrag: a database built for the exploration of bioactive fragment space for drug discovery. J Chem Inf Model 58(9):1725–1730
Kolb P, Caflisch A (2006) Automatic and efficient decomposition of two-dimensional structures of small molecules for fragment-based high-throughput docking. J Med Chem 49:7384–7392
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. In Chem Biol (pp. 243–250). Humana Press, New York, NY
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):1–13
Hess B, Kutzner C, Van Der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4(3):435–447
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65(3):712–725
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25(2):247–260
Sousa da Silva AW, Vranken WF (2012) ACPYPE-antechamber python parser interface. BMC Res Notes 5(1):1–8
Berendsen HJ, Postma JP, van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration. In Intermolecular forces (pp. 331–342). Springer, Dordrecht
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126(1):014101
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52(12):7182–7190
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J Chem Phys 98(12):10089–10092
Hess B, Bekker H, Berendsen HJ, Fraaije JG (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18(12):1463–1472
Kumari R, Kumar R, Open Source Drug Discovery Consortium & Lynn A (2014) g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962
Aqvist J, Marelius J (2001) The linear interaction energy method for predicting ligand binding free energies. Comb Chem High Throughput Screening 4(8):613–626
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Cheatham TE (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Gilson MK, Honig B (1988) Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins 4(1):7–18
Almlöf M, Brandsdal BO, Åqvist J (2004) Binding affinity prediction with different force fields: examination of the linear interaction energy method. J Comput Chem 25(10):1242–1254
Carlson HA, Jorgensen WL (1995) An extended linear response method for determining free energies of hydration. J Phys Chem 99(26):10667–10673
Loida PJ (1994) Molecular specificity of substrate recognition and activation in cytochrome P-450 (CAM). Doctoral dissertation, University of Illinois at Urbana-Champaign
Bakan A, Meireles LM, Bahar I (2011) ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27(11):1575–1577
Bakan A, Bahar I (2009) The intrinsic dynamics of enzymes plays a dominant role in determining the structural changes induced upon inhibitor binding. Proc Natl Acad Sci 106(34):14349–14354
Yang L, Song G, Carriquiry A, Jernigan RL (2008) Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure 16(2):321–330
Eyal E, Yang LW, Bahar I (2006) Anisotropic network model: systematic evaluation and a new web interface. Bioinformatics 22(21):2619–2627
Maisuradze GG, Liwo A, Scheraga HA (2009) Principal component analysis for protein folding dynamics. J Mol Biol 385(1):312–329
Ross C, Nizami B, Glenister M, Sheik Amamuddy O, Atilgan AR, Atilgan C, Tastan Bishop Ö (2018) MODE-TASK: large-scale protein motion tools. Bioinformatics 34(21):3759–3763
Ertl P, Rohde B, Selzer P (2000) Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 43(20):3714–3717
Daina A, Michielin O, Zoete V (2014) iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model 54(12):3284–3301
Potts RO, Guy RH (1992) Predicting skin permeability. Pharm Res 9(5):663–669
Acknowledgements
JP and LD thank the UGC Non-NET fellowship, and PA thanks the UGC for Junior Research Fellowship. All authors thank CMSD, University of Hyderabad, for providing computational facilities.
Author information
Authors and Affiliations
Contributions
LGP and DL perceived the research problem. PA, JP, and DL carried out the work. PA, JP, and DL wrote the manuscript. LGP corrected the manuscript. All authors approve the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This manuscript does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Andola, P., Pagag, J., Laxman, D. et al. Fragment-based inhibitor design for SARS-CoV2 main protease. Struct Chem 33, 1467–1487 (2022). https://doi.org/10.1007/s11224-022-01995-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-01995-z