
Abstract
Two Chlamydomonas reinhardtii mutants defective in CHLM encoding Mg-protoporphyrin IX methyltransferase (MgPMT) were identified. The mutants, one with a missense mutation (chlM-1) and a second mutant with a splicing defect (chlM-2), do not accumulate chlorophyll, are yellow in the dark and dim light, and their growth is inhibited at higher light intensities. They accumulate Mg-protoporphyrin IX (MgProto), the substrate of MgPMT and this may be the cause for their light sensitivity. In the dark, both mutants showed a drastic reduction in the amounts of core proteins of photosystems I and II and light-harvesting chlorophyll a/b-binding proteins. However, LHC mRNAs accumulated above wild-type levels. The accumulation of the transcripts of the LHC and other genes that were expressed at higher levels in the mutants during dark incubation was attenuated in the initial phase of light exposure. No regulatory effects of the constitutively 7- to 18-fold increased MgProto levels on gene expression were detected, supporting previous results in which MgProto and heme in Chlamydomonas were assigned roles as second messengers only in the transient activation of genes by light.
Similar content being viewed by others


Introduction
The unicellular green alga Chlamydomonas reinhardtii has genetic and physiological features that make it an ideal eukaryotic photosynthetic model organism that differs from monocotyledonous and dicotyledonous plants. Its plastid, mitochondrial, and nuclear genomes have been sequenced and procedures for the transformation of all 3 genomes have been elaborated (Grossman et al. 2004; Merchant et al. 2007; Rochaix 1995; 2002). These characteristics as well as the alga’s potential for genetic analyses facilitate the identification of the function of genes. Unlike angiosperms, C. reinhardtii can metabolize an organic carbon source (acetate), enabling the alga to grow heterotrophically in the dark. The chloroplasts in these dark-grown cells contain functional thylakoid membranes including the photosynthetic chlorophyll-binding proteins of photosystems (PS) I and II.
The eukaryotic chlorophyll biosynthetic pathway leads to chlorophylls a and b (Fig. 1). Each enzymatic intermediate is well-defined, and enzymatic activities and genes have been detected and described for many steps of the pathway (Beale 1999; Moulin and Smith 2005). Although generally quite similar, the pathways for tetrapyrrole biosynthesis in vascular plants and C. reinhardtii show some distinct differences. For example, heme in the green alga appears to be synthesized exclusively in plastids. Only single genes encoding protoporphyrinogen (Protogen) oxidase and ferrochelatase were found in the C. reinhardtii genome and the corresponding gene products were localized to the chloroplast, suggesting that C. reinhardtii possesses only one single heme-synthesizing pathway located in this organelle (van Lis et al. 2005). Tobacco and cucumber were shown to have two genes for each of these enzymatic steps (Fig. 1) allowing the parallel synthesis of heme in plastids and mitochondria (Lermontova et al. 1997; Suzuki et al. 2002). Also, unlike angiosperms but similar to gymnosperms, C. reinhardtii cells have both a light-independent protochlorophyllide oxidoreductase (POR) consisting of three proteins (ChlN, ChlB, ChlL) encoded by the chloroplast genome, and a light-dependent POR (Fig. 1). C. reinhardtii mutants defective in the light-independent conversion of protochlorophyllide to chlorophyllide are yellow in the dark but, due to the presence of a light-dependent POR, are green in the light. Defects in at least 7 nuclear loci have been shown to cause a yellow-in-the-dark mutant phenotype (designated y-1 and y-5 to y-10). These loci are required for the production of a functional light-independent POR enzyme (Cahoon and Timko 2000).

Scheme of the tetrapyrrole biosynthetic pathway in Chlamydomonas reinhardtii and vascular plants. Shown are the major intermediates and the genes mentioned in the text. Dashed lines indicate multiple steps
Presently, several concepts for tetrapyrrole-mediated signaling have been proposed, which reflects differences in organisms, plant development, genotype, and growth conditions (Beck and Grimm 2006; Kropat et al. 1997,2000; Nott et al. 2006; Papenbrock et al. 2000; Woodson and Chory 2008). In Arabidopsis treated with the carotenoid synthesis inhibitor norflurazon, MgProto had been assigned an essential role in the repression of Lhcb1 (Strand et al. 2003). However, this interpretation was put in question when the reported accumulation of MgProto under these conditions was not observed by others (Mochizuki et al. 2008; Moulin et al. 2008). As alternative plastid signaling factors controlling Lhcb1 expression in carotenoid-deficient plants exposed to light, ROS or an altered redox state of the plastids were suggested (Kleine et al. 2009; Mochizuki et al. 2008).
In C. reinhardtii a tetrapyrrole-derived signaling pathway was elaborated based on the observation that feeding of MgProto, MgProtoMe, or heme transiently induced a subset of nuclear genes including HEMA (encoding glutamyl-tRNA reductase) and chaperone genes HSP70A and HSP70B (Kropat et al. 1997; Vasileuskaya et al. 2005; von Gromoff et al. 2008). Since the feeding experiments were performed in the dark, a role of ROS in signaling could be excluded. This same set of genes is also transiently induced by shifting cell cultures from dark to light (Kropat et al. 1997; Vasileuskaya et al. 2005; von Gromoff et al. 2008). A transient, light-induced increase in MgProto and/or MgProtoMe appears to be a prerequisite for the activation of these genes by light (Kropat et al. 2000). However, since an accumulation of these tetrapyrroles in dark-grown cells did not activate gene expression, we postulated that light in addition is required to make the plastid-produced tetrapyrroles accessible to downstream signal transduction components in the cytosol/nucleus (Kropat et al. 2000; Beck 2005). The hypothesis that MgProto, MgProtoMe, and heme are second messengers in this light-signaling pathway is supported by the characterization of an enhancer element in the promoter regions of these genes that mediates the induction by both the tetrapyrroles and light (von Gromoff et al. 2006).
The isolation of mutants that accumulate MgProto may provide an independent test for this hypothesis. Using the ability of C. reinhardtii to grow in darkness with acetate as sole source of carbon and energy, we identified two mutants that accumulate excessive amounts of MgProto. These mutant clones are yellow and do not green in the light. Thus, they are different from the y mutants with defects in light-independent protochlorophyllide reduction. The two mutants have lesions in the CHLM gene, which encodes Mg-protoporphyrin IX methyltransferase (MgPMT).
Materials and methods
Algal strains and culture conditions
Chlamydomonas reinhardtii wild-type strain CC-124 was obtained from the Chlamydomonas Culture Collection at the University of Minnesota, and wild-type strain 4A+ (mt+) was provided by J.-D. Rochaix (University of Geneva). The mutants were generated in the 4A+ strain by UV mutagenesis applying between 30 and 60 mJ cm−2. After mutagenesis the cells were plated and incubated in the dark. Mutant clones were recognized by their altered pigmentation.
The ΔpetA, F15, and F35 mutants were provided by O. Vallon (Institut de Biologie Physico-Chimique, Paris). ΔpetA is a deletion mutant strain lacking the chloroplast petA gene encoding cytochrome f. The mutant also was shown to have strongly reduced levels of the Rieske protein (Kuras and Wollman 1994). The PS I mutant F15 is deficient in TAB 1, a gene required for translation of psaB mRNA. F15 is unable to synthesize PsaA and PsaB, two PS I reaction center polypeptides (Girard et al. 1980; Stampacchia et al. 1997). The F35 mutant has a nuclear mutation that causes a defect in the translation of the chloroplast psbA mRNA, which encodes D1, a core polypeptide of PS II (Yohn et al. 1996). Mutants defective in D1 are unable to assemble a stable PSII complex (Bennoun et al. 1986). If not otherwise noted, strains were grown heterotrophically in Tris-Acetate-Phosphate (TAP) medium (Harris 1989) on a rotatory shaker at 23°C either in the dark or irradiated with white light at the fluence rates indicated. For selection of photoautotrophic growth, the same medium without acetate (TMP) was employed. Mutant stocks were maintained on TAP agar medium in the dark. Light induction was performed according to Kropat et al. (1995) by irradiation with white light provided by fluorescent tubes (Osram L36W/25).
Nuclear transformation of C. reinhardtii
Chlamydomonas reinhardtii nuclear transformation was performed using the glass bead method (Kindle 1990) modified as described previously (Kropat et al. 2000). In a typical transformation assay, 108 cells, 100 ng of plasmid DNA or 1 μg of BAC DNA were used. Immediately after vortexing with glass beads, cells were spread onto freshly prepared TMP plates (1% agar). Plates were incubated at 23°C in the light (~60 μmol photons m−2 s−1). Transformants that complemented the mutations could be detected as green colonies. Transformants were collected and analysed after 3 weeks.
RNA gel blot analyses
Total RNA was isolated from 20 ml cultures grown to 2–4 × 106 cells per ml. The procedures employed for RNA extraction, separation on agarose gels, and blotting were as described previously (von Gromoff et al. 1989), except that the nylon membranes used were Hybond-N (Amersham). The probes used for hybridization were as follows: HSP70A (a 3.8-kb SalI cDNA fragment; von Gromoff et al. 1989), HSP70B (a 2.1-kb EcoRI/BamHI cDNA fragment; von Gromoff et al. 1989), CHLH (a 1.4-kb EcoRI cDNA fragment; Chekounova et al. 2001; Vasileuskaya et al. 2004), CHLD (a 680-bp PCR fragment; Vasileuskaya et al. 2004), CRD-1 (a 1.2-kb XhoI, PstI fragment; Moseley et al. 2000), LHCBM6 (a 2.4-kb EcoRI/HindIII genomic fragment; Hahn and Kück 1999), LHCBM2 (a ~1.5-kb PstI, NcoI cDNA fragment; Vasileuskaya et al. 2004), LHCBM1 (a 400-bp AatII, SacI cDNA fragment; Vasileuskaya et al. 2004), HEM15 (a 338-bp PCR fragment: F-Primer: 5′AAGCTCGACGACGTCAAGCC3′, R-Primer: 5′TACAGCGGGAGGATGAC CAG3′), ALAD (a ~1.7-kb cDNA fragment; Matters and Beale 1995), HEMA (a 560-bp PCR fragment; Vasileuskaya et al. 2004), CHLM (a 248-bp PCR fragment: F-Primer: 5′CAGTGCCTAAACACCAAGCCT3′; R-Primer:5′TTCTGAGGGTCAGTAGCACA3′). The CHLM probe is homologous to exon I. For CHLI we used a 510-bp EcoRI, BamHI cDNA fragment derived from gene CHLI1. This probe has 78% sequence identity to the corresponding region of CHLI2, a second gene for the I subunit in the genome of C. reinhardtii (Grossman et al. 2004), and thus is expected to hybridize to CHLI2 mRNA as well. CBLP, which encodes a Gβ-like polypeptide (a 1-kb EcoRI genomic fragment; von Kampen et al. 1994) was used as loading control. Plasmids containing fragments of HEMA and ALAD were kindly provided by Prof. Beale (Providence). Probes were labeled with 50 μCi [α-32P]dCTP (Amersham) according to the random priming protocol (Feinberg and Vogelstein 1983). Hybridizations were performed as described previously (Wegener and Beck 1991). After hybridization the membranes were washed twice in 2× SSC for 5 min at room temperature, once in 2× SSC, 1% SDS for 30 min at 65°C, and once in 0.2× SSC for 30 min at room temperature. RNA gel blots were screened by exposing membranes to BAS-MP imaging plates (Fuji). They were evaluated by a phosphor imager (Bio-Rad Laboratories). For the quantitative determination of changes in mRNA abundance the Quantity One 4.5.1 program from Bio-Rad Laboratories was used.
Isolation of CHLM
BAC clones containing CHLM were identified by screening the membrane-immobilized DNA of a C. reinhardtii BAC library (Lefebvre and Silflow 1999) obtained from Clemson University Genomics Institute (http://www.genome.clemson.edu/) using the CHLM probe described above. Hybridization of the membranes was performed using the protocol provided by the Clemson University Genomics Institute. Positive BAC clones (37J5, 34G12, 3G11, 33E3) that contained the CHLM gene were obtained from Clemson University Genomics Institute. DNA was isolated using the following protocol: 100 ml 2×YT medium (1.6% w/v bactotryptone; 1% w/v bacto yeast extract; 0.5% w/v NaCl) with 12.5 μg/ml chloramphenicol were inoculated with bacteria harboring BAC DNA and incubated overnight at 37°C. Cells were harvested by centrifugation at 4000 rpm, 5 min (GSA rotor, Sorvall) and resuspended in 5 ml TES buffer (10 mM Tris–HCl, pH 8; 1 mM EDTA; 100 mM NaCl). The suspension was incubated at room temperature for 5 min and 5 ml lysis solution (200 mM NaOH; 1% SDS) were added. The suspension was mixed gently and incubated on ice for 5 min. 5 ml Buffer I (3 M potassium acetate, 5 M acetic acid) were added and gently mixed by inversion. Incubation on ice was continued for at least 5 min. The cell debris was pelleted by centrifugation at 10000 rpm for 15 min. To the supernatant 5 μl 10 mg/ml RNase were added followed by incubation at 37°C for 30 min. 15 ml PCI (50% v/v phenol; 49% v/v chloroform; 1% v/v isoamylalcohol) were then added to the tube, mixed by inverting and centrifuged at 10000 rpm for 15 min at room temperature (SA-600 rotor, Sorval). The aqueous layer was transferred to a new tube and 15 ml isopropanol were added to precipitate DNA for 1 h at room temperature. DNA was pelleted by centrifugation at 10000 rpm for 30 min at room temperature and the pellet was washed 2× with 70% ethanol. The dry pellet was dissolved in 400 μl TE buffer and transferred into a 1.5 ml Eppendorf tube. One volume PCI was added to the tube, mixed by inverting and centrifuged at 10°C for 10 min. The aqueous layer was transferred to a 1.5 ml Eppendorf tube. DNA was precipitated with 2.5 volumes 96% ethanol, 1/10 volume 3 M Na acetate (pH 5.3) and pelleted by centrifugation (10000 rpm, 20 min at room temperature). The DNA pellet was washed with 70% ethanol, dried and dissolved in TE buffer or water. By digestion of the BAC DNA with different enzymes (Eco57I, AatII, Bsp1407I, NheI, RsaI) and Southern blot analyses it was confirmed that the ordered BAC clones contained the complete CHLM gene.
Cloning and sequencing of CHLM
Genomic sequence data (http://genome.jgi-psf.org/Chlre3/Chlre3.home.html) provided information on restriction sites used for cloning of CHLM. A 6315-bp fragment containing the entire CHLM gene was obtained by digestion of DNA of BAC clone 3G11 with BbvCI. The fragment, after blunt ending, was ligated into the EcoRV site of pBluescriptSK+/− (Stratagene). Clones containing inserts of correct size were verified by partial sequencing and restriction analysis. DNA of one clone was tested for its ability to complement the mutants defective in chlorophyll synthesis upon transformation.
For the identification of the mutations in two mutants that could be complemented by the CHLM gene, DNA was extracted from both mutant strains using the CTAB method (von Gromoff et al. 1989). The sequence of the CHLM mutant alleles was determined after amplification of overlapping fragments that comprised the entire CHLM coding region, using four primer pairs. The upstream (A) and downstream (B) primers were as follows: 1A, 5′-GTGAGTGGCAAAGAGTGCT-3′; 1B, 5′-TGTTCACCTCGTCCGTCTC-3′; 2A, 5′-ACTACTTCAACACTGCCGG-3′; 2B, 5′-CCGCCACCGCCTGCTGGT-3′; 3A, 5′-TGGCGTCTGAGGCGGAGCAG-3′; 3B 5′-TGCGCTTGAGGATGCTGTACG-3′; 4A, 5′-TCATCATCTCGTTCGCACC-3′; 4B, 5′-CACGCTAGGCACACGCTTC-3′. The reactions were performed for 35 cycles of 1 min at 95°C, 1 min at 63°C, and 2.5 min at 72°C with Pfu DNA polymerase (Fermentas), which possesses proofreading activity. Resulting PCR products were subcloned into vector pGEM-T (Promega) and sequenced. Mutated sites were confirmed by sequencing of independently derived clones. Sequencing was performed by the dideoxynucleotide chain termination method (Sanger et al. 1977) using the ALF DNA analyzing system (Amersham Biosciences Europe).
RT–PCR for the identification of RNA products that result from the mutation in chlM-2
A OneStep RT–PCR kit was used for RT–PCR following the instructions provided by the manufacturer (Qiagen) using the following primers: F-Primer: 5′ATGGCGTCTGAGGCGGAGCA3′ (located in exon 4); R-Primer: 5′CGGGGAACAGCTCGCCAATG3′ (located in exon 6).
Pigment analysis
Porphyrin steady-state levels were determined in cells grown in liquid in the dark. Cells were collected and stored at −80°C prior to rupture by sonication in an ice bath. Porphyrins were extracted in three steps with (1) methanol, (2) potassium-phosphate buffer, and (3) a mixture of acetone: methanol: 0.1 Ν NH4OH (10:9:1, v/v/v) (Alawady and Grimm 2005). Aliquots of the supernatant were separated by HPLC (model 1100; Agilent) on a RP 18 column (Novapak C18, 4-μm particle size, 3.9 × 150 mm; Waters Chromatography) at a flow rate of 1 ml/min in a methanol/0.1 M ammonium acetate, pH 5.2, gradient and eluted with a linear gradient of solvent B (90% methanol and 0.1 M ammonium acetate, pH 5.2) and solvent A (10% methanol and 0.1 M ammonium acetate, pH 5.2). The eluted samples were monitored by a fluorescence detector at an excitation wavelength of 405 nm and emission wavelength of 620 nm for Proto and an excitation wavelength of 420 nm and emission wavelength of 595 nm for Mg-porphyrins. The porphyrins and metalloporphyrins were identified and quantified by comparison with authentic standards purchased from Frontier Scientific. The content of noncovalently bound heme was determined after removal of chlorophyll from C. reinhardtii cells by intensive washing with ice-cold alkaline acetone containing 0.1 N NH4OH (9:1, v/v) (Weinstein and Beale 1984). Heme was then extracted with acidic acetone containing 5% HCl, transferred to diethyl ether, concentrated, and washed on a DEAE-Sepharose column. Absorbance was monitored at 398 nm using a millimolar extinction coefficient of 144. The concentration of heme standard solutions was determined spectrophotometrically (Weinstein and Beale 1983).
Analysis of carotenoid contents
Dark-grown cells were used for the analysis of carotenoids. Cells were harvested and stored at −80°C prior to rupture by sonication in an ice bath. Carotenoids were analyzed essentially as described by Gilmore and Yamamoto (1991). Separation was performed on a HPLC system (Agilent-1100) using 25-cm columns from Waters (Lichrosphere 100RP-18). The separation of carotenoids was achieved by a linear gradient, employing a mixture of 89.9% acetonitrile, 10% ddH2O, 0.1% amine, pH 8.0 and 100% ethylacetate at a flow rate of 1 ml/min. The outflow was monitored by a photodiode array detector (Agilent). Carotenoid standards were purchased from Roth and used for quantification.
Protein extraction and immunoblot analyses
For protein isolation, 10-ml cell suspensions were harvested by centrifugation and resuspended in 300 μl 0.1 M Na2CO3, 0.1 M dithiothreitol. After adding 200 μl 5% SDS 30% sucrose cells were broken by incubation for 45 s at 100°C. Protein concentrations were determined with amido black (Popov et al. 1975). After separation of whole-cell proteins by 14% SDS–polyacrylamide gel electrophoresis (Laemmli 1970), proteins were transferred to polyvinylidene difluoride membranes (Hybond-P; Amersham) by semidry blotting using a discontinuous transfer system, according to the manufacturer’s recommendations (Bio-Rad). Transfer was performed at 0.8 mA per cm2. Blocking and immunodecorations were performed in phosphate buffered saline containing 3% nonfat dry milk. Immunodetection was done by the enhanced chemiluminescence technique according to the manufacturer’s protocol (Amersham), using anti-rabbit immunoglobulin G linked to horseradish peroxidase as the secondary antibody (Sigma). Antisera for specific detection were against HSP70A [kindly provided by J. Rosenbaum (Yale)], HSP70B (Schroda et al. 1999), and MgPMT. The polyclonal MgPMT antibody (produced by Seq-lab, Göttingen) was raised against a recombinant MgPMT, isolated by affinity purification from E. coli extracts. The antisera against LHCII, LHC, CP43, CP47 (de Vitry et al. 1989), cytochrome f (Pierre and Popot 1993) and Rieske (de Vitry et al. 1989) were kindly provided by F.-A. Wollman (IBPC, Paris). PsaA antiserum was from Agrisera. The antiserum for PsaD was kindly provided by M. Hippler (University of Muenster, Germany). CHLH antiserum was from R. Willows, CRD1 antiserum was kindly provided by S. Merchant (UCLA). Antisera directed against the β subunit of plastidic ATP synthase (CF1) were kindly provided by O. Vallon (IBPC, Paris).
Determination of ALA synthesizing capacity
Chlamydomonas reinhardtii cells were grown in 200 ml of liquid TAP medium to a density of 3–5 × 106 cells per ml. The cells were harvested, collected by centrifugation, resuspended and incubated in TAP medium, pH 7.1, that contained 20 mM levulinic acid. The incubation time was 10 h; then the cells were collected by centrifugation and frozen in liquid nitrogen. Cell samples were resuspended in 1 ml 20 mM potassium phosphate buffer, pH 6.8, and centrifuged. The ALA assays and quantification of ALA-pyrroles with Ehrlich reagent were performed as described by Mauzerall and Granick (1956).
Determination of enzyme activities
Chlamydomonas reinhardtii cells were grown in 2 l of liquid TAP medium to a density of 3–5 × 106 cells per ml. Cells were collected by centrifugation, disrupted by sonication in cold homogenization buffer (0.5 M sorbitol, 0.1 M Tris–HCl, 1 mM DTT, 0.1% BSA, pH 7.5), and subdivided for assays of Mg-chelatase, MgPMT and ferrochelatase activity which were performed according to Alawady et al. (2005).
CHLM gene model
The CHLM gene model is found in the Chlamydomonas genome sequence (http://genome.jgi-psf.org/Chlre3/Chlre3.home.html) (CHLM gene model: #RWI_chlre3.78.3.1.1; protein ID 195575).
Results
Identification of mutants defective in CHLM
To identify C. reinhardtii mutants defective in chlorophyll synthesis, wild-type cells were subjected to UV-mutagenesis, grown in the dark and screened for a chlorophyll-free phenotype. Based on the initial assumption of a defect in chlorophyll biosynthesis, Proto and Mg-porphyrin levels were determined. Two yellow mutants were selected as potentially defective in MgPMT activity because they accumulated excessive amounts of MgProto, the substrate of the enzyme. In the C. reinhardtii genome a single gene encoding MgPMT (CHLM) has been identified by sequence comparison with CHLM genes from other organisms (Lohr et al. 2005). To identify the mutant loci, we first identified BAC clones containing the predicted CHLM gene. DNA of these BAC clones was employed in transformation experiments. Complemented mutants produced green colonies capable of photo-autotrophic growth. These complementation tests were subsequently repeated with a subcloned BbvCI DNA-fragment of about 6300 bp, predicted to contain the CHLM gene. The two mutants complemented by the CHLM gene-containing DNA fragment were named chlM-1 and chlM-2.
Characterization of the mutations
The structure of the C. reinhardtii CHLM gene, as deduced from combined genomic and EST sequence data (http://genome.jgi-psf.org/Chlre3/Chlre3.home.html) is presented in Fig. 2a. This gene, interrupted by 6 introns, encodes a protein with a high degree of similarity to MgPMT of higher plants and cyanobacteria. The overall amino acid identity of C. reinhardtii MgPMT was 62% with Nicotiana tabacum, 58% with Arabidopsis thaliana, 57% with Oryza sativa, 54% with Synechocystis, and 26% with Rhodobacter capsulatus. A plastid localization of C. reinhardtii MgPMT is supported by a transit peptide of 52 amino acids predicted by the ChloroP program (Emanuelsson et al. 1999).

Structure of the CHLM gene, alterations caused by the mutations chlM-1 and chlM-2, and test of the mutants for CHLM encoded RNA and protein. a Gene CHLM with 7 exons. Open boxes indicate deduced protein coding exons, black boxes indicate 5′ and 3′ untranslated regions, and lines indicate introns. The positions and sequence alterations of mutations chlM-1 and chlM-2 are indicated. b Genomic and RT–PCR sequences in the vicinity of the splice site between exon 4 and intron 4. The vertical arrows indicate the end of exon 4. The bases of intron 4 are indicated in italics. The altered base of intron 4 in mutant chlM-2 is shown in bold. The asterisk indicates a stop codon. c For the determination of CHLM mRNA levels, samples were taken from wild type and mutant cultures grown in the dark. RNA gel blot hybridizations were performed using specific probes for CHLM and CBLP, the latter serving as a loading control. The size of CHLM mRNAs was deduced by comparison with an RNA marker. The average levels of CHLM mRNA in the mutants relative to the wild type (corrected for differences in loading) ±SEM are given below the RNA blot. For details see Materials and methods. Cultures for protein extraction were grown with low intensity irradiation (15 μmol m−2 s−1). After SDS–PAGE separation of 60 μg of total soluble protein per slot and blotting to nitrocellulose membranes, MgPMT was detected by immunodecoration with MgPMT-specific antibodies. For a loading control, the same membrane was incubated with antibodies directed against the 1β subunit of the chloroplast ATPase (CF1β)
The mutations in the gene were identified by sequencing PCR fragments generated with genomic DNA or RT–PCR fragments of the mutants. In the chlM-1 mutant, a T to C transition mutation was discovered in exon 6 which is predicted to result in a replacement of a leucine by a proline residue at position 271 (Fig. 2a). Analysis of mutant chlM-2 revealed a G to A transition at the first base of intron 4 (Fig. 2a). RT–PCR analysis employing primers homologous to exon 4 and exon 6 revealed that mRNA from chlM-2 yielded one product of about 900 bp indicating a defect in the splicing reaction at intron 4 (data not shown). In this unspliced RNA an in-frame stop codon (TGA) is predicted right after the 3’ end of exon 4 (Fig. 2b) leading to a predicted unprocessed protein with 218 aa instead of 326 aa of wild-type MgPMT.
Total RNA of dark-grown cultures was subjected to RNA blot analysis using a CHLM-specific probe to assess the amount of CHLM mRNA in the two mutants. Both wild type and the chlM-1 mutant produced CHLM mRNA of the same size (about 1400 bases) and similar quantity (Fig. 2c). In contrast, the CHLM mRNA of mutant chlM-2 was about 600 bases bigger. This increase in size of the mutant transcript may be accounted for by the presence of intron 4 (622 bp). The amount of chlM-2 mutant mRNA was reproducibly and significantly elevated (Fig. 2c). Evidently, a rapid degradation by nonsense-mediated mRNA decay does not occur. The protein complex required for this RNA decay is recruited to mRNA during nuclear RNA splicing (Chang et al. 2007). The absence of a splicing reaction at the border between exon 4 and intron 4 thus may prevent nonsense-mediated RNA decay in the chlM-2 mutant. Whether this absence also accounts for a longer half-life of the chlM-2 mRNA remains to be elucidated.
To assess the consequences of the mutations at the level of the MgPMT protein, polyclonal MgPMT-specific antiserum was raised. The serum detected a gene product with a mass of about 28 kDa that correlates well with an expected MW of 29700 for processed MgPMT. A protein of the same size was also observed in mutant chlM-1 (Fig. 2c). However, no immune-reacting protein band in the MW region expected for the truncated gene product was observed in mutant chlM-2. The absence of an immune-reacting polypeptide suggests either instability of the truncated gene product or an inhibition of its synthesis in mutant chlM-2.
Levels of chlorophyll, heme, tetrapyrrole intermediates, and carotenoids are affected in the mutants
To analyse the consequences of the two mutations in CHLM we determined pool levels of tetrapyrroles and chlorophyll. Only residual levels of chlorophyll were detected in both mutants (Table 1). The missense mutant (chlM-1) had reproducibly somewhat higher contents than chlM-2. Methyl transferases of low specificity may be responsible for the residual formation of MgProtoMe and thus chlorophyll in MgPMT-defective mutants (Grimm, unpublished data) and, in the case of chlM-1, a low activity of the mutant protein can not be excluded. MgProtoMe steady state levels were reduced and the levels of Proto showed only a modest increase (maximally twofold in chlM-2) (Table 1). The pool levels of the MgPMT substrate MgProto were elevated 7- to 18-fold. For comparison, C. reinhardtii mutants defective in Mg-chelatase exhibit at least tenfold increases in Proto levels (Chekounova et al. 2001; von Gromoff et al. 2008). The levels of non-covalently bound heme in both mutants were reduced about twofold, although the substrate of ferrochelatase (Proto) was present at elevated levels. The difference to Mg-chelatase mutants which, in comparison to wild type, exhibit 2- to 5-fold elevated heme levels (von Gromoff et al. 2008) suggests a different regulation of heme synthesis in MgPMT- and Mg-chelatase-deficient mutants. Thus, mutants defective in Mg-chelatase and MgPMT, although catalyzing sequential steps in chlorophyll biosynthesis, exhibit distinctly different effects on the intracellular concentration of Proto, MgProto and heme.
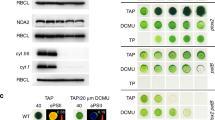
The two chlM mutants in the dark exhibited yellow pigmentation, very similar to that of the well studied y (yellow in the dark) mutants (Cahoon and Timko 2000).These y mutants have yellow colour in the dark, but are green in the light (Fig. 3). In contrast to the y mutants, the chlM mutants stayed yellow also upon exposure to low light. In addition, at medium light intensities (45 μmol m−2 s−1), growth of the chlM-2 mutant was inhibited (Fig. 3) and higher light intensities (500 μmol m−2 s−1) were required to inhibit growth of chlM-1. The increased light sensitivity of chlM-2 (Fig. 3) correlates with a greater severity of the mutation and about threefold higher levels of MgProto compared to chlM-1 (Table 1).

Growth and pigmentation of the two chlM mutants. The wild- type strain as well as a yellow-in-the-dark mutant (y7) are shown for control. After inoculation the plates were incubated for 15 days either in the dark, or irradiated with the fluence rates indicated
The yellow pigmentation of the mutants, uncovered by the virtual absence of chlorophyll, may be explained by the presence of carotenoids. As shown in Table 2, the concentration of β-carotene observed in the two chlM mutants was about the same as that of wild type. However, contents of xanthophyll cycle pigments were distinctly reduced in the mutants (Table 2). These data revealed that the block in chlorophyll synthesis significantly affects xanthophyll accumulation.
Light-regulated expression of CHLM in wild type and mutants
We next investigated whether in mutants chlM-1 and chlM-2 the steady-state and light-inducible expression of CHLM at the RNA and protein levels were affected. Accumulation of CHLM mRNA was induced by a shift of wild-type and mutant cultures from dark to light. Due to the light sensitivity of the mutants only a fluence rate of 15 μmol m−2 s−1 was employed (Fig. 4a). In the two mutants, CHLM mRNA accumulated to higher levels than in wild type (in the case of chlM-2 the RNA concentration at time point 0 already was elevated as shown in Fig. 2). This mRNA accumulation was maintained for a longer time period.

Light induction of CHLM in wild type and mutants. a mRNA accumulation in cultures that after growth in the dark were shifted into dim light (15 μmol m−2 s−1). Samples were taken at the time points indicated (hours). RNA gel blots were performed as described in Materials and methods using specific probes for CHLM and CBLP, the latter serving as a loading control. The average fold induction of mRNA relative to the dark control (corrected for differences in loading) from 9 independent experiments ±SEM were determined. b Changes in MgPMT upon shift of cultures from dark to light. For the assay of MgPMT, total soluble protein was extracted from cultures grown in the dark and after exposure to light (15 μmol m−2 s−1) for one or 2 h. After blotting, the proteins were detected by immunodecoration with MgPMT-specific antibodies. The weak signal seen in chlM-2 mutant extracts may reflect an unspecific reaction since it was not seen in Fig. 1c and also does not increase upon light incubation. For a loading control, the same membrane was decorated with an antiserum directed against the chloroplast ATPase subunit CF1β
A distinct increase in MgPMT was seen 2 h after transfer to light in wild type and chlM-1 (Fig. 4b). Both in wild type and chlM-1, a second protein band that migrated slower than the one present in dark-grown cells, was seen already one h after shift to light; after 2 h this protein appeared to be the dominant species. It likely represents MgPMT after posttranslational modification. The cause for this change in gel migration of MgPMT upon exposure of cells to light is the subject of separate studies.
Mutations in CHLM have reverse effects on LHC proteins and LHC mRNA levels
The accumulation of chlorophyll binding proteins may be controlled by the availability of chlorophyll at the levels of gene expression and/or protein turnover. To distinguish between these possibilities we made use of the ability of C. reinhardtii to grow heterotrophically in the dark when supplied with acetate. Exposure to light of mutant cells that accumulate intermediates of tetrapyrrole biosynthesis may cause ROS production and, consequently, a diverse array of responses (op den Camp et al. 2003; Shao et al. 2007).
The antibodies available to us detected four LHC proteins in the wild type, but no or only very low amounts of protein were observed in the mutants defective in CHLM (Fig. 5a). We next assayed for the presence of three specific LHC mRNAs in these dark-grown cultures. As illustrated in Fig. 5b, the steady-state LHC mRNA levels were significantly elevated in the mutants when compared to the parental strain. The most pronounced accumulation (~tenfold) was observed for LHCBM1 mRNA. Thus, in the MgPMT-defective mutants, an upregulation of the LHC genes was observed while, at the same time, the LHC proteins were nearly absent. Evidently, the mutations have a divergent effect on LHC gene expression: enhanced transcription or stabilization at the mRNA level and reduced accumulation of LHC proteins due to downregulation at the translational level and/or a decrease in protein stability as a result of chlorophyll deficiency.

Immuno and RNA blot analyses of genes coding for components of the photosynthetic apparatus in mutants defective in CHLM. a Detection of proteins of the light harvesting complex. Cultures of the strains prior to harvest were grown in darkness for at least 3 days. Each lane of the SDS-gel received the same amount of protein. We used an antiserum that specifically detected the LHCII protein and one that reacted with 3 different light harvesting proteins. Antisera that detected the CF1β protein of plastid ATPase served as a loading control. b mRNA abundance of three genes encoding light harvesting chlorophyll a/b binding proteins. Cultures were grown in the dark and RNA was isolated, processed, and hybridized with the gene-specific probes indicated. The quantitative evaluation of the RNA gel blots comprised at least 4 independent experiments. The average mRNA levels relative to those of the wild type (set as 1) and corrected for differences in loading ±SEM are given. CBLP served as a loading control. c Detection of selected proteins of PSI, PSII, and the cytb 6 /f complex. Besides extracts of the chlM mutants and wild type also those from mutants with defined lesions were employed for control: mutant F15 is deficient in the synthesis of PsaB (62), mutant F35 does not produce protein D1 (Yohn et al. 1996), and mutant ΔpetA has an inactivated petA (cytochrome f) gene (Kuras and Wollman 1994)
Effect of chlM mutations on photosystem I, II, and the cytb 6 /f complex proteins
The absence of LHC proteins in the chlM mutants suggested that the accumulation of proteins of the photosystems and the cytochrome b 6 /f complex might be affected as well. Indeed, in the two chlM mutants PSI core proteins PsaA and PsaD were strongly reduced or absent, respectively (Fig. 5c). A test for PSII core proteins CP43 and CP47 also revealed their absence in both mutants (Fig. 5c). As expected, presence of chlorophyll is a prerequisite for the assembly or stability of PSI and PSII subunits. In contrast, the concentration of two core proteins of the cytb 6 /f complex, cytochrome f and the Rieske Fe–S protein, was similar in the chlM mutants to that seen in the parental strain (Fig. 5c).
In chlM mutants a subset of genes exhibits moderately altered expression patterns
To analyze the effect of the mutations in CHLM on gene expression, we assayed for the steady-state mRNA levels of a selected set of genes in wild type and mutant cultures grown in the dark. Mg-chelatase, a heterotrimeric enzyme composed of subunits H, D and I, plays a crucial role in the channelling of Proto into the chlorophyll branch (Beck and Grimm 2006). In comparison to wild type both chlM mutants revealed a pronounced increased amount of Mg-chelatase H subunit (Fig. 6a). Elevated subunit H levels correlated with increased levels of CHLH mRNA (Fig. 6b). 1.7- and 1.9-fold increased mRNA levels were also observed for subunits I and D, respectively.

Immuno and RNA blot analyses of genes coding for enzymes of chlorophyll synthesis. a Levels of proteins involved in chlorophyll biosynthesis of wild type and chlM mutants cultivated in the dark. Each slot of the SDS-gel received the same amount of protein. Immunodecoration was done using antisera against CHLH and CRD1, the latter detecting subunits of the cyclase (Moseley et al. 2002). Antisera that detect the CF1β protein of plastid ATPase served as a loading control. b Comparison between chlM mutants and wild type in expression levels of genes involved in tetrapyrrole biosynthesis. mRNA steady state levels of CHLH, CHLI, CHLD, CRD-1, HEMA (encoding glutamyl-tRNA reductase), ALAD (encoding ALA dehydratase) and HEM15 (encoding Fe-chelatase) in wild type and mutant cultures. c Steady state levels of mRNA of HSP70 genes encoding a cytosolic (HSP70A) and a plastidic (HSP70B) chaperone. Cultures were grown in the dark. RNA was isolated, processed, and hybridized with the gene-specific probes indicated. The quantitative evaluation of the RNA gel blots comprised at least three independent experiments. The average mRNA levels relative to those of the wild type (set as 1) and corrected for differences in loading ±SEM are given. CBLP served as a loading control
The cyclase that converts MgProtoMe to divinyl protochlorophyllide is a multimeric complex (Rzeznicka et al. 2005). Two immune-reacting bands (Cth1 and Crd1) which are components of this complex are recognized by the Crd1 antibody (Moseley et al. 2002). The slower migrating immune-reacting protein presumably is Cth1(Fig. 6a), which is encoded by a gene not repressed by the presence of copper in the medium, while the faster migrating protein may represent Crd1, which is encoded by a gene repressed by copper (Moseley et al. 2002). Apparently, both proteins are present in dark-grown cells and their concentration appears not to be affected significantly by the mutations in CHLM. At the CRD1 mRNA level, 1.6- to 1.9- fold increases were detected in the two mutants when compared to wild type (Fig. 6b).
Genes encoding earlier steps in tetrapyrrole biosynthesis [HEMA encodes glutamyl-tRNA reductase, ALAD encodes 5-amino levulinic acid (ALA) dehydratase, HEM15 encodes ferrochelatase] exhibited only minor variations in mRNA steady state levels when the chlM mutants were compared with wild type (Fig. 6b).
Analysis of the mRNA levels of light-inducible genes HSP70A and HSP70B revealed elevated levels for both genes in dark-grown mutant cultures (Fig. 6c).
Assay of enzyme activities crucial for tetrapyrrole biosynthesis
We next tested whether mutations in CHLM affect the activities of enzymes involved in tetrapyrrole biosynthesis and whether the variations seen at the mRNA or protein levels correlate with enzyme activity. No activity for MgPMT was detected in chlM-1 and chlM-2, which either still expresses or lacks the MgPMT protein, respectively (Fig. 7). These data suggest that the amino acid exchange in chlM-1 resulted in a loss of MgPMT function—a result supported by the detection of only low amounts of chlorophyll in this mutant (Table 1). The reduced Mg-chelatase activity observed in both mutants as compared to wild type (Fig. 7) is in contrast to the elevated H-subunit levels (Fig. 6a). This suggests that the assembly of Mg-chelatase is impaired and/or one of the other two subunits (I or D) was limiting for enzyme activity. Alternatively, posttranslational mechanisms inactivated the Mg-chelatase complex in response to lack of functional MgPMT. Indeed, Mg-chelatase has been reported to be regulated by interaction with thioredoxin (Ikegami et al. 2007) and the GUN4 protein (Larkin et al. 2003). Also ferrochelatase activity in the mutants was reduced (Fig. 7). This reduction in enzyme activity correlates with the lower levels of noncovalently bound heme observed in the mutants as compared to the parental strain (Table 1). The capacity for the synthesis of ALA by the first 2 enzymes of tetrapyrrole biosynthesis was shown to be elevated in the mutants. Mutant chlM-2, harbouring the stronger mutant allele, shows a higher increase in ALA biosynthesis rate (Fig. 7). It is suggested that the slightly increased levels of HEMA expression contribute to the elevated ALA-synthesizing capacity. In addition, the increase in ALA-synthesizing capacity may be attributed to a reduced feedback inhibition of glutamyl-tRNA reductase by heme (Vothknecht et al. 1998) which in the mutants was reduced more than twofold (Table 1).

Activities of selected enzymes for heme and chlorophyll biosynthesis. The specific activities in dark-grown cells of wild type and two chlM mutants were determined for MgPMT, Mg-chelatase, ferrochelatase, and for the 5-aminolevulinic acid (ALA) synthesizing enzymes. The activities were determined as described in Materials and methods. The average of 3 independent experiments ±SEM are given. In the case of ALA synthesis the average of 2 independent experiments which did not differ by more than 10% is presented
Test for light inducibility of selected genes
A comparative analysis of changes in expression patterns at the mRNA level induced by a shift of wild-type and mutant cultures from dark to light revealed that light induction of all genes analyzed was maintained in chlM-2 (Fig. 8 and Supplementary Fig. S1) and chlM-1 (Supplementary Fig. S1). The degree and kinetics of mRNA accumulation observed exhibited no significant changes for the majority of genes that are involved in tetrapyrrole biosynthesis (HEMA, ALAD, CRD-1, HEM-15) when chlM-2 and chlM-1 (Supplementary Fig. S1) were compared to wild type. In contrast, the mRNAs for the three subunits of Mg-chelatase (CHLH, CHLD, and CHLI) in the mutants showed a distinctly retarded accumulation when compared to wild type, i.e., 2- to 4-fold lower mRNA levels in the 1 h samples (Fig. 8a and Supplementary Fig. S1). A similar observation was made with the LHC genes tested: The initial time course of mRNA accumulation in the mutants was distinctly attenuated after transition from dark to light when it was compared to wild type, i.e., 1.5- to 2.8-fold lower mRNA levels in the 2 h samples (Fig. 8b and Supplementary Fig. S1). After 4 h of light treatment the degree of accumulation observed is ultimately similar in both mutants and wild type. A delayed mRNA accumulation in the first hours after onset of light was also observed for HSP70A and HSP70B: mRNA accumulation was more than twofold reduced in the 1 h sample in both mutants in comparison to wild type (Fig. 8c and Supplementary Fig. S1). Clearly, in both allelic mutants patterns of RNA accumulation are similar and distinct from those of the wild type control, supporting an influence of defects in CHLM on light induction of the genes tested.

Effect of mutation chlM-2 on gene expression after a shift from dark to light. Cultures of wild type and the chlM-2 mutant grown in the dark at time 0 were shifted into dim light (15 μmol m−2 s−1) and samples for RNA isolation were taken at the time points indicated (hours). The average levels of mRNA accumulation relative to the dark control and corrected for differences in loading ±SEM from at least 3 independent experiments are indicated. CBLP served as a loading control. a Test for light induction of Mg-chelatase genes. b Test for light induction of genes encoding light harvesting chlorophyll a/b binding proteins. c Test for light induction of HSP70 genes
Discussion
Two mutants with yellow pigmentation have been characterized that contained excessive amounts of MgProto and were deficient in chlorophyll. These two mutants did not green in the light; they remained yellow at low fluence rates, and showed inhibition of growth at higher light intensities (Fig. 3). Growth was more severely inhibited in chlM-2, which is characterized by higher levels of MgProto, than in chlM-1, suggesting that toxic products of MgProto photooxidation are responsible. The genetic defects in the two mutants could be assigned to CHLM, which encodes MgPMT, an enzyme essential for chlorophyll biosynthesis (Fig. 1). The yellow pigmentation of the mutants is mainly accounted for by β-carotene that is present at wild type levels (Table 2). β-carotene levels in dark-grow y mutants lacking chlorophyll, were previously shown to be similar to those of light-grown chlorophyll containing y mutants. The bulk of this carotenoid is found in the eyespot globules of this organism (Goldberg and Ohad 1970). In contrast to β-carotene, levels of carotenoids with oxygenated endgroups (xanthophylls) in the mutants were strongly reduced (Table 2). These xanthophylls are constituents of the light-harvesting antenna pigment protein complexes (Kühlbrandt et al. 1994; Niyogi et al. 1997). As a consequence of chlorophyll deficiency, accumulation of xanthophyll pigments, and stability of LHC apoproteins are compromised in the MgPMT-defective mutants.
The defects in CHLM in both mutants were caused by point mutations. While in chlM-2, due to an RNA splicing defect, no MgPMT was detected (Fig. 2c), in chlM-1 a replacement of leucine at position 271 by a proline residue is predicted to inactivate and possibly to destabilize MgPMT (Fig. 2). The mutated site is in a conserved region of MgPMT, which is predicted to have an α-helical structure that extends from amino acid 265 to amino acid 276 (Protein Structure Prediction Server, http://bioinf.cs.ucl.ac.uk/psipred/). This region of the protein is part of the classical motif of S-adenosyl-methionine-dependent methyltransferases (Block et al. 2002).
Consequences of MgPMT deficiency on enzymes at the branch point of chlorophyll synthesis and ALA synthesis
The absence of MgPMT correlates with a reduced activity of both Mg-chelatase and ferrochelatase (Fig. 7). Although an increased expression of CHLH in the mutants was seen at both the RNA and the protein level (Fig. 6), it did not manifest itself in higher Mg-chelatase activity (Fig. 7). Similar observations for CHLH expression were made in an Arabidopsis MgPMT-deficient mutant (Pontier et al. 2007). The reduced Mg-chelatase activity observed may be accounted for by a lack of interaction between the H-subunit of Mg-chelatase with MgPMT in the mutants (Alawady et al. 2005). Apparently, both proteins belong to a protein complex that catalyzes the early steps of Mg-porphyrin biosynthesis and thus ensure efficient metabolic flux (Hinchigeri et al. 1997; Shepherd et al. 2005). Also in transgenic CHLM antisense tobacco plants with lowered levels of CHLM mRNA and MgPMT, reduced Mg-chelatase activity was observed (Alawady and Grimm 2005). Vice versa, in mutants defective in Mg-chelatase of tobacco and C. reinhardtii a reduction in MgPMT activity was seen (Alawady, unpublished data). However, in contrast to the situation in C. reinhardtii, a lowering of MgPMT in tobacco resulted in a reduction in Mg-chelatase H-subunit mRNA and protein (Alawady and Grimm 2005) suggesting that the underlying regulatory mechanisms are different. The catalytic slow-down of Mg-chelatase in lines lacking MgPMT might, however, also result from the accumulation of MgProto at the product site of Mg-chelatase.
The upregulation in the rate-limiting 5-aminolevulinic acid (ALA) synthesis observed in dark-grown C. reinhardtii chlM mutants (Fig. 7) is similar to that observed for Proto-accumulating C. reinhardtii mutants impaired in Mg-chelatase subunit H (Chekounova et al. 2001). In contrast, in vascular plants, the enzymes at the beginning of the Mg-porphyrin branch exert a tight negative feedback regulation on the activity of the ALA-synthesizing pathway (Papenbrock et al. 2000; Alawady and Grimm 2005). This feedback-control was demonstrated as response to reduced expression of genes for individual Mg-chelatase subunits as well as by lower and over-expressed transcript levels of the gene encoding MgPMT in transgenic tobacco plants. Lower activity of Mg-chelatase and MgPMT in tobacco did not result in an accumulation of Proto and MgProto, respectively, but lowered ALA synthesis rates. The feedback control likely operates at the transcriptional and posttranslational levels (Alawady and Grimm 2005; Papenbrock et al. 2000). We speculate that this feedback system did not evolve in C. reinhardtii due to the ability of this alga to reduce protochlorophyllide in the dark and thus to experience to a lower extent the risk of adverse effects of accumulation of porphyrins and Mg-porphyrins.
Effect of mutations in CHLM on components of the thylakoidal electron transport chain
Defects in CHLM result in the reduction of PSI and PSII core components as well as light harvesting proteins, often to levels below detection limits (Fig. 5a, c). This absence of apoproteins most likely is a consequence of the strong reduction in chlorophyll levels. When this pigment is not synthesized, the chlorophyll-binding proteins are not stabilized in thylakoids. Parallel synthesis of chlorophyll and carotenoids is required for correct assembly of chlorophyll binding proteins and their insertion into the photosynthetic complexes in thylakoid membranes (Paulsen 2001). Protein complexes lacking chlorophyll become substrates for proteases (Herrin et al. 1992; Eichacker et al. 1996).
We did not observe in both mutants any modification in the contents of cytochrome f and Rieske protein, two core components of the cytb 6 /f complex (Fig. 5c). Previously, a C. reinhardtii mutant blocked in chlorophyll synthesis was shown not to accumulate the cytb 6 /f complex (Pierre et al. 1997). Also in a chlorophyll-deficient Arabidopsis mutant harbouring a knockout of CHLM, cytochrome f was not detected (Pontier et al. 2007). Our results suggest that the residual chlorophyll levels observed in the mutants (chlM-1: 5%, chlM-2: 1.5%) are sufficient for a stable assembly of the complex. The preferential routing of residual chlorophyll into this complex may be explained by the high affinity of the cytb 6 /f complex for chlorophyll (Pierre et al. 1997).
Consequences of mutations in CHLM on signaling and the regulation of gene expression
The most striking aberration in gene expression observed is the upregulation of LHC mRNA levels in both mutants (Fig. 5b). Remarkably, it is accompanied by the simultaneous absence of light harvesting proteins (Fig. 5a). The upregulation of LHC mRNA levels observed here is unlikely to be triggered by the elevated MgProto levels in the mutants since these genes were shown not to respond to this tetrapyrrole (Vasileuskaya et al. 2004) and, in addition, long term activation was shown to result in an inactivation of the signaling pathway downstream from MgProto (von Gromoff et al. 2008). In a CHLM knockout mutant of Arabidopsis and in tobacco lines expressing a CHLM antisense construct, a downregulation of Lhcb mRNA levels has been observed (Alawady and Grimm 2005; Pontier et al. 2007). This downregulation in the case of the Arabidopsis mutant was interpreted as a consequence of the elevated MgProto pools in this mutant (Pontier et al. 2007). However, this interpretation was put in question by the analysis of a different MgPMT-deficient Arabidopsis mutant that, although it accumulates a large amount of MgProto, still expressed Lhcb1 at levels equivalent to wild type. For the downregulation of Lhcb1 observed previously the photosensitizing effect of the accumulated MgProto, i.e., the production of ROS, was suggested to be responsible (Mochizuki et al.2008). The molecular explanation for the higher LHC mRNA levels in C. reinhardtii chlM mutants is an interesting topic for future investigation.
Light induction of the genes tested is clearly maintained in both MgPMT-deficient mutants (Fig. 4, Fig. 8, Supplementary Fig. S1). These data suggest that the lack of chlorophyll or the accumulation of intermediates of tetrapyrrole biosynthesis does not interfere with the basic mechanisms of light induction at low intensities. The signaling routes by which light activates these genes differ: The LHC genes, ALAD, as well as the Mg-chelatase genes are controlled by the blue light receptor phototropin (Im et al. 2006). No information on the signaling components that mediate light activation of CHLM, HEM15 and CRD-1 are available. In the case of HSP70A, HSP70B, and possibly HEMA, MgProto and heme have been suggested to act as second messengers (Kropat et al. 1997, 2000; Vasileuskaya et al. 2005; von Gromoff et al. 2008). Here, no effect of elevated Mg-porphyrin pool levels (Table 1) on light-induced mRNA accumulation is detected (Fig. 8). This is consistent with the observation that irradiation by itself already causes maximal mRNA accumulation that cannot be surpassed by the feeding of MgProto to the culture medium (Kropat et al. 1997, 2000; von Gromoff et al. 2006, 2008).
A closer inspection of the time course data revealed that those genes upregulated in dark-grown cultures of the two mutants, i.e., the HSP70, LHC, and CHLH, CHLI, and CHLD genes, showed a distinct attenuation in the initial time course of RNA accumulation when mutants where compared to wild type (Fig. 8, Supplementary Fig. S1). This complex response suggests that gene expression is monitored via feedback regulation independently from the pathway which mediates the induction, i.e., the genes are subject to multiple layers of regulation.
An involvement of MgProto in the regulation of those genes that exhibit elevated expression in dark grown cultures is unlikely for the following reasons. The 7- to 18-fold elevated levels of MgProto observed in the mutants (Table 1) resulted only in a relatively small increase in the expression of genes (HSP70A, HSP70B, HEMA) that previously have been shown to be regulated by MgProto (Fig. 6). We have reported previously that a light-induced ~fivefold increase in MgProto resulted in a more than sixfold increase in HSP70A, HSP70B, and HEMA mRNA levels (Kropat et al. 1997, 2000; Vasileuskaya et al. 2005; von Gromoff et al. 2008). The minor effect of MgProto in the chlM mutants in the dark is consistent with previous data on the regulatory role of this tetrapyrrole: MgProto, its methyl ester, and heme have been implicated as second messengers in transient gene activation after a shift of cultures from dark to light. Constitutively elevated levels of MgProto, MgProtoMe, or heme in dark-grown cultures are not expected to have a signaling function for two reasons: (a) Increased MgProto and MgProtoMe levels generated by the feeding of Proto to cultures in the dark did not manifest themselves in HSP70A activation (Kropat et al. 2000). We postulated that irradiation of the cultures was required to make these chloroplast-synthesized tetrapyrroles accessible to factors of the downstream signaling pathway in cytosol/nucleus. (b) The signaling pathway that mediates gene activation by MgProto and heme is turned off upon continuous activation (von Gromoff et al. 2008). Hence, the analysis of gene expression in the chlM mutants with elevated tetrapyrrole pools supports the hypothesis that MgProto, MgProtoMe, and heme only mediate the transient activation of genes in response to a light signal.
On the other hand, the basis for the observed upregulation of tetrapyrrole biosynthetic genes can be explained by disturbances caused by the lack of MgPMT activity. It is important to keep in mind that our assays of steady-state levels of mRNA, protein, and enzyme activities were performed with cultures grown continuously in the dark. Any differences in gene expression thus are not caused by artifactual responses elicited by irradiation of mutant cultures. Changes in regulatory patterns between mutants and wild type instead may be assigned to a limited number of differences: lack of chlorophyll, alterations in steady-state levels of tetrapyrroles (other than MgProto, MgProtoMe or heme), and/or disturbance of enzyme activities at the branch point between heme and chlorophyll synthesis. Which of these factors affect gene expression remains to be elucidated.
For the chaperone genes HSP70A and HSP70B we favour the idea that their elevated mRNA levels in the mutants (Fig. 6) are due to an increased demand for molecular chaperones caused by the turnover of proteins that, in the absence of chlorophyll, are not stably assembled. The increased sequestering of chaperones by substrates is known to activate HSP gene expression via the autoregulatory heat-shock-factor system (Morimoto 2002). These studies of gene expression patterns in the MgProto accumulating mutants with defects in MgPMT are in agreement with a function of this tetrapyrrole in the transient activation of genes that follows upon exposure of dark-adapted cultures to light.
References
Alawady AE, Grimm B (2005) Tobacco Mg-protoprophyrin IX methyl-transferase is involved in inverse activation of Mg-porphyrin and protoheme synthesis. Plant J 41:282–290
Alawady A, Reski R, Yaronskaya E, Grimm B (2005) Cloning and expression of the tobacco CHLM sequence encoding Mg-protoporphyrin IX methyltransferase and its interaction with Mg-chelatase. Plant Mol Biol 57:679–691
Beale SI (1999) Enzymes of chlorophyll biosynthesis. Photosynth Res 60:43–73
Beck CF (2005) Signaling pathways from the chloroplast to the nucleus. Planta 222:743–756
Beck CF, Grimm B (2006) Involvement of tetrapyrroles in cellular regulation. In: Grimm B, Porra RJ, Rüdiger W, Scheer H (eds) Biochemistry, biophysics and biological functions of chlorophylls. Springer, Dordrecht, pp 223–233
Bennoun P, Spierer-Herz M, Erickson J, Girard-Bascou J, Pierre Y, Delosme M, Rochaix J-D (1986) Characterization of photosystem II mutants of Chlamydomonas reinhardtii lacking the psbA gene. Plant Mol Biol 6:151–160
Block MA, Teward AK, Albrieux C, Joyard EM, Joyard J (2002) The plant S-adenosyl-L-methionine: Mg-protoporphyrin IX methyltransferase is located in both envelope and thylakoid chloroplast membranes. Eur J Biochem 269:240–248
Cahoon AB, Timko MP (2000) Yellow-in-the-dark mutants of Chlamydomonas lack the CHLL subunit of light-independent protochlorophllide reductase. Plant Cell 12:559–568
Chang YF, Imam JS, Wilkinson MF (2007) The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76:51–74
Chekounova E, Voronetskaya V, Papenbrock J, Grimm B, Beck CF (2001) Characterization of Chlamydomonas mutants defective in the H-subunit of Mg-chelatase. Mol Genet Genomics 266:363–373
de Vitry C, Olive J, Drapier D, Recouvreur M, Wollman FA (1989) Posttranslational events leading to the assembly of photosystem II protein complex: a study using photosynthesis mutants from Chlamydomonas reinhardtii. J Cell Biol 109:991–1006
Eichacker LA, Helfrich M, Rüdiger W, Müller B (1996) Stabilization of chlorophyll a-binding apoproteins P700, CP47, D2, and D1 by chlorophyll a or Zn-pheophytin a. J Biol Chem 271:32174–32179
Emanuelsson O, Nielsen H, von Heijne G (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8:978–984
Feinberg AP, Vogelstein B (1983) A technique for radiolabelling DNA restriction endonuclease fragments to high activity. Anal Biochem 132:2–13
Gilmore AM, Yamamoto HY (1991) Zeaxanthin formation and energy-dependent fluorescence quenching in pea chloroplasts under artificially mediated linear and cyclic electron transport. Plant Physiol 96:635–643
Girard J, Chua NH, Bennoun P, Schmidt G, Delosme M (1980) Studies in mutants deficient in the photosystem I reaction centers in Chlamydomonas reinhardtii. Curr Genet 2:215–221
Goldberg I, Ohad I (1970) Biogenesis of chloroplast membranes. J Cell Biol 44:563–571
Grossman AR, Lohr M, Im CS (2004) Chlamydomonas reinhardtii in the landscape of pigments. Annu Rev Genet 38:119–173
Hahn D, Kück U (1999) Identification of DNA sequences controlling light- and chloroplast-dependent expression of the lhcb1 gene from Chlamydomonas reinhardtii. Curr Genet 34:459–466
Harris EH (1989) The Chlamydomonas Source Book: A comprehensive guide to biology and laboratory use. Academic Press, San Diego
Herrin DL, Battey JF, Greer K, Schmidt GW (1992) Regulation of chlorophyll apoprotein expression and accumulation. J Biol Chem 267:8260–8269
Hinchigeri SB, Hundle B, Richards WR (1997) Demonstration that the BchH protein of Rhodobacter capsulatus activates S-adenosyl-L-methionine: magnesium protoporphyrin IX methyltransferase. FEBS Lett 407:337–342
Ikegami A, Yoshimura N, Motohashi K, Takahashi S, Romano PGN, Hisabori T, Takamiya K, Masuda T (2007) The CHLI1 subunit of Arabidopsis thaliana magnesium chelatase is a target protein of the chloroplast thioredoxin. J Biol Chem 282:19282–19291
Im CS, Eberhard S, Huang K, Beck CF, Grossman AR (2006) Phototropin involvement in the expression of genes encoding chlorophyll and carotenoid biosynthesis enzymes and LHC apoproteins in Chlamydomonas reinhardtii. Plant J 48:1–16
Kindle KL (1990) High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 87:1228–1232
Kleine T, Voigt C, Leister D (2009) Plastid signalling to the nucleus: messengers still lost in the mists? Trends Genet 25:185–192
Kropat J, von Gromoff ED, Müller FW, Beck CF (1995) Heat shock and light activation of a Chlamydomonas HSP70 gene are mediated by independent regulatory pathways. Mol Gen Genet 248:727–734
Kropat J, Oster U, Rüdiger W, Beck CF (1997) Chlorophyll precursors are signals of chloroplast origin involved in light induction of nuclear heat-shock genes. Proc Natl Acad Sci USA 94:14168–14172
Kropat J, Oster U, Rüdiger W, Beck CF (2000) Chloroplast signalling in the light induction of nuclear HSP70 genes requires the accumulation of chlorophyll precursors and their accessibility to cytoplasm/nucleus. Plant J 24:523–531
Kühlbrandt W, Wang DN, Fujiyoshi Y (1994) Atomic model of plant light-harvesting complex by electron crystallography. Nature 367:614–621
Kuras R, Wollman FA (1994) The assembly of cytochrome b 6 /f complexes: an approach using genetic transformation of the green alga Chlamydomonas reinhardtii. EMBO J 13:1019–1027
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Larkin MR, Alonso JM, Ecker JR, Chory J (2003) GUN4, a regulator of chlorophyll synthesis and intracellular signaling. Science 299:902–906
Lefebvre PA, Silflow CD (1999) Chlamydomonas: The cell and its genome. Genetics 151:9–14
Lermontova I, Kruse E, Mock HP, Grimm B (1997) Cloning and characterization of a plastidal and a mitochondrial isoform of tobacco protoporphyrinogen IX oxidase. Proc Natl Acad Sci USA 94:8895–8900
Lohr M, Im CS, Grossman AR (2005) Genome-based examination of chlorophyll and carotenoid biosynthesis in Chlamydomonas reinhardtii. Plant Physiol 138:490–515
Matters GL, Beale SI (1995) Structure and expression of the Chlamydomonas reinhardttii alaD gene encoding the chlorophyll biosynthetic enzyme, delta-aminolevulinic acid dehydratase (porphobilinogen synthase). Plant Mol Biol 27:607–617
Mauzerall D, Granick S (1956) The occurrence and determination of delta-amino-levulinic acid and porphobilinogen in urine. J Biol Chem 219:435–446
Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Whitman GB, Terry A, Salamov A, Fritz-Laylin LK, Maréchal-Drouard L, Marshall WF, Qu LH, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren Q, Ferris P, Lindquist E, Shapiro H, Lucas SM, Grimwood J, Schmutz J, Chlamydomonas Annotation Team, JGI Annotation Team, Grigoriev IV, Rokhsar DS, Grossman AR (2007) The Chlamydomonas genome reveals the evolution of the key animal and plant functions. Science 318:245–251
Mochizuki N, Tanaka R, Tanaka A, Masuda T, Nagatani A (2008) The steady-state level of Mg-protoporphyrin IX is not a determinant of plastid-to-nucleus signaling in Arabidopsis. Proc Natl Acad Sci USA 105:15184–15189
Morimoto RI (2002) Dynamic remodeling of transcription complexes by molecular chaperones. Cell 110:281–284
Moseley J, Quinn J, Eriksson M, Merchant S (2000) The Crd1 gene encodes a putative di-iron enzyme required for photosystem I accumulation in copper deficiency and hypoxia in Chlamydomonas reinhardtii. EMBO J 19:2139–2151
Moseley JL, Page MD, Alder NP, Ericksson M, Quinn J, Soto F, Theg SM, Hippler M, Merchant S (2002) Reciprocal expression of two candidate di-iron enzymes affecting photosystem I and light-harvesting complex accumulation. Plant Cell 14:673–688
Moulin M, Smith AG (2005) Regulation of tetrapyrrole biosynthesis in higher plants. Biochem Soc Trans 33:737–742
Moulin M, McCormac AC, Terry MJ, Smith AG (2008) Tetrapyrrole profiling in Arabidopsis seedlings reveals that retrograde plastid nuclear signaling is not due to Mg-protoporphyrin IX accumulation. Proc Natl Acad Sci USA 105:15178–15183
Niyogi KK, Björkman O, Grossman AR (1997) The roles of specific xanthophylls in photoprotection. Proc Natl Acad Sci USA 94:14162–14167
Nott A, Jung HS, Koussevitzky S, Chory J (2006) Plastid-to-nucleus retrograde signaling. Annu Rev Plant Biol 57:739–759
op den Camp RG, Przybya D, Ochsenbein C, Laloi C, Kim C, Danon A, Wagner D, Hideg E, Göbel C, Feussner I, Nater M, Apel K (2003) Rapid induction of distinct stress responses after the release of singlet oxygen in Arabidopsis. Plant Cell 15:2320–2332
Papenbrock J, Mock HP, Tanaka R, Kruse E, Grimm B (2000) Role of magnesium chelatase activity in the early steps of the tetrapyrrole biosynthetic pathway. Plant Physiol 122:1161–1169
Paulsen H (2001) Pigment assembly: transport and ligation. In: Aro EM, Andersson B (eds) Advances in photosynthesis, vol 9, regulatory aspects of photosynthesis. Kluwer Academic Publishers, Dordrecht, pp 219–233
Pierre Y, Popot JL (1993) Identification of two 4-kDa miniproteins in the cytochrome b 6 f complex from Chlamydomonas reinhardtii. C R Acad Sci III 316:1404–1409
Pierre Y, Breyton C, Lemoine Y, Robert B, Vernotte C, Popot JL (1997) On the presence and role of a molecule of chlorophyll a in the cytochrome b 6 f complex. J Biol Chem 272:21901–21908
Pontier D, Albrieux C, Joyard J, Lagrange T, Block MA (2007) Knock-out of the magnesium protoporphyrin IX methyltransferase gene in Arabidopsis. Effects on chloroplast development and on chloroplast-to-nucleus signaling. J Biol Chem 282:2297–2304
Popov N, Schmitt S, Matthices H (1975) Eine störungsfreie Mikromethode zur Bestimmung des Proteingehalts in Gewebshomogenaten. Acta Biol Germ 31:1441–1446
Porra RJ, Grimme LH (1974) A new procedure for the determination of chlorophylls a and b and its application to normal and regreening Chlorella. Anal Biochem 57:255–267
Rochaix JD (1995) Chlamydomonas reinhardtii as the photosynthetic yeast. Annu Rev Genet 29:209–230
Rochaix JD (2002) The three genomes of Chlamydomonas. Photosynth Res 73:285–293
Rzeznicka K, Walker CJ, Westergren T, Kannangara CG, von Wettstein D, Merchant S, Gough SP, Hansson M (2005) Xantha-I encodes a membrane subunit of the aerobic Mg-protoporphyrin IX monomethyl ester cyclase involved in chlorophyll biosynthesis. Proc Natl Acad Sci USA 102:5886–5891
Sanger F, Nicklen S, Coulson AR (1977) DNA sequence with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Schroda M, Vallon O, Wollman FA, Beck CF (1999) A chloroplast-targeted heat shock protein 70 (HSP70) contributes to the photoprotection and repair of photosystem II during and after photoinhibition. Plant Cell 11:1165–1178
Shao N, Krieger-Liszkay A, Schroda M, Beck CF (2007) A reporter system for the individual detection of hydrogen peroxide and singlet oxygen: its use for the assay of reactive oxygen species produced in vivo. Plant J 50:475–487
Shepherd M, McLean S, Hunter CN (2005) Kinetic basis for linking the first two enzymes of chlorophyll biosynthesis. FEBS J 272:4532–4539
Stampacchia O, Girard-Bascou J, Zanasco JL, Zerges W, Bennoun P, Rochaix JD (1997) A nuclear-encoded function essential for translation of the chloroplast psaB mRNA in Chlamydomonas. Plant Cell 9:773–782
Strand A, Asami T, Alonso J, Ecker JR, Chory J (2003) Chloroplast to nucleus communication triggered by accumulation of Mg-protoporphyrinIX. Nature 421:79–83
Suzuki T, Masuda T, Singh DP, Tan FC, Tsuchiya T, Shimada H, Ohta H, Smith AG, Takamiya K (2002) Two types of ferrochelatase in photosynthetic and nonphotosynthetic tissues of cucumber: their difference in phylogeny, gene expression, and localization. J Biol Chem 277:4731–4737
van Lis R, Atteia A, Nogaj LA, Beale SI (2005) Subcellular localization and light-regulated expression of protoporphyrinogen IX oxidase and ferrochelatase in Chlamydomonas reinhardtii. Plant Physiol 139:1946–1958
Vasileuskaya Z, Oster U, Beck CF (2004) Involvement of tetrapyrroles in inter-organellar signaling in plants and algae. Photosynth Res 82:289–299
Vasileuskaya Z, Oster U, Beck CF (2005) Identification of Mg-protoporphyrin IX and heme as plastid/mitochondrial factors that control HEMA expression in Chlamydomonas. Eukaryot Cell 4:1620–1628
von Gromoff ED, Treier U, Beck CF (1989) Three light-inducible heat shock genes of Chlamydomonas reinhardtii. Mol Cell Biol 9:3911–3918
von Gromoff ED, Schroda M, Oster U, Beck CF (2006) Identification of a plastid response element that acts as an enhancer within the Chlamydomonas HSP70A promoter. Nucl Acids Res 34:4767–4779
von Gromoff ED, Alawady A, Meinecke L, Grimm B, Beck CF (2008) Heme, a plastid-derived regulator of nuclear gene expression in Chlamydomonas. Plant Cell 20:552–567
von Kampen J, Nieländer U, Wettern M (1994) Stress-dependent transcription of a gene encoding a Gβ-like polypeptide from Chlamydomonas reinhardtii. J Plant Physiol 143:756–758
Vothknecht UC, Kannangara CG, von Wettstein D (1998) Barley glutamyl tRNAGlu reductase: mutations affecting haem inhibition and enzyme activity. Phytochem 47:513–519
Wegener D, Beck CF (1991) Identification of novel genes specifically expressed in Chlamydomonas reinhardtii zygotes. Plant Mol Biol 16:937–946
Weinstein JD, Beale SI (1983) Separate physiological roles and subcellular compartments for two tetrapyrrole biosynthetic pathways in Euglena gracilis. J Biol Chem 258:6799–6807
Weinstein JD, Beale SI (1984) Biosynthesis of protoheme and heme as precursors solely from glutamate in the unicellular red alga Cyanidium caldarium. Plant Physiol 74:146–151
Woodson JD, Chory J (2008) Coordination of gene expression between organellar and nuclear genomes. Nat Rev Genet 9:383–395
Yohn CB, Cohen A, Danon A, Mayfield SP (1996) Altered mRNA binding activity and decreased translation initiation in a nuclear mutant lacking translation of the chloroplast psbA mRNA. Mol Cell Biol 16:3560–3566
Acknowledgments
Erika D. von Gromoff’s expert technical assistance is greatly appreciated. We thank Martin Lohr (Universität Mainz) for helpful suggestions and a gift of loroxanthin. This work was supported by grants from the Deutsche Forschungsgemeinschaft to C.F.B. (Be 903/13-2) and B.G. (Gr 936/14-1).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Meinecke, L., Alawady, A., Schroda, M. et al. Chlorophyll-deficient mutants of Chlamydomonas reinhardtii that accumulate magnesium protoporphyrin IX. Plant Mol Biol 72, 643–658 (2010). https://doi.org/10.1007/s11103-010-9604-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-010-9604-9