
Abstract
Acylated flavonoids are widely distributed natural dietary bioactives with several health attributes. A large diversity of acylated flavonoids with interesting biological potentialities were reported. Of these, 123 compounds with potential antimicrobial, antiparasitic, anti-inflammatory, anti-nociceptive, analgesic and anti-complementary effects were selected from several databases. Based upon these data, the possible mechanistic evidence for their effects were reported. Generally, aromatic acyls i.e., galloyl derivatives appeared to improve efficacy through enhancement of the binding affinities to molecular targets due to plenty of donating and accepting centers. Docking simulations conducted by Molecular Operating Environment (MOE) of acylated flavonoids revealed that compound 12 is at the top of the list into the antibacterial target DNA gyrase subunit B (GyrB), from E. coli, followed by compounds 10, 4 and 23. Compounds 81, 88, 96, 92, 99, 100, 102 and 103 have the strongest binding affinities into Human matrix metallopeptidase (MMP) 2 and 9 catalytic domains. Compound 103 exerted the most balanced predicted dual MMP-2/MMP-9 inhibition action. Compound 95 recorded the strongest binding affinity into metabotropic glutamate receptor (mglur1) with the lowest energy conformer. The data presented in this review suggests that these candidate acylated flavonoids ought to be considered in future drug developments especially as anti-inflammatory and antimicrobial agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant kingdom comprises a myriad of secondary metabolite classes that are crucial for plant growth, defense mechanism against invaders as well as tolerance towards biotic and abiotic types of stresses (Wang et al. 2019). The major secondary metabolite classes found in plants are alkaloids, terpenes, steroids, and phenolics, with flavonoids found almost ubiquitous in all plants (Kumar and Pandey 2013; Teles et al. 2018). Generally, the diversity among secondary metabolites including flavonoid subclasses arises from structural modifications on the basic skeleton as hydroxylation, methylation, acylation and glycosylation, and to further affect physicochemical properties, stability, solubility and bioavailability (Biely et al. 2014; Viskupičová et al. 2009). As per flavonoids class, modifications of the basic skeletons usually occur in plants through tailored enzymatic reactions. Flavonoids are polyphenols in nature, with 8000 flavonoids reported in literature (de Araújo et al. 2017; Wang et al. 2019).
Interest in dietary flavonoids is attributed to their positive role against various ailments such as cardiovascular risks, cancer, inflammations, and microbial and viral infections (Ardhaoui et al. 2004; de Araújo et al. 2017). Although flavonoids are well recognized for their medicinal effects, yet their applications are rather limited owing to restrictions in their solubility and stability, with polar groups to lower their binding to cell membranes and cellular targets (Ardhaoui et al. 2004; de Araújo et al. 2017; Viskupičová et al. 2009). Chemical modification of the flavonoid structure such as acylation could thus strongly influence their biological activities through improved stability, bioavailability and lipophilic cell membrane penetration and permeability (Biely et al. 2014; Sudan and Rupasinghe 2015).
In plants, flavonoids acylation occurs through two main routes aromatic or aliphatic acylation. The aromatic acylation commonly includes the addition of galloyl, feruloyl, p-coumaroyl or caffaeoyl moieties to the parent molecule and this leads to stabilization of flavonoid molecule or intensifying the color as in case of anthocyanins pigmentation (Abe et al. 2008). As described above, the presence of the aliphatic substituent as acetyl, malonyl or long chain acyl moieties improved its stability (Biely et al. 2014; Viskupičová et al. 2009).
Owing to a major role played by inflammatory processes in different diseases, a great interest has been paid for controlling these processes using natural products such as flavonoids. Recent studies have revealed that flavonoids can modify enzymes and transcription factors important for controlling inflammatory mediators (Maleki et al. 2019). Previous studies reported on flavonoids such as diosmetin, hesperetin, kaempferol and astragalin to improve their anti-inflammatory activities 30 times upon acylation (de Araújo et al. 2017; Harborne and Williams 2000). Matrix metalloproteinases (MMPs) are 32 zinc-dependent structurally and functionally related endopeptidases in humans that are implicated in extracellular matrix degradation and remodeling, with a myriad of vital physiological processes (Sternlicht and Werb 2001; Nagase et al. 2006; Page-McCaw et al. 2007). MMPs dysfunction leads to several pathological conditions ranging from inflammation, osteoarthritis, multiple sclerosis, autoimmune diseases to cancer (Roy et al. 2009; Whittaker et al. 1999). Considering inflammation, MMPs family, especially MMP-2 and -9, have been reported to influence inflammation cascade in a myriad of action mechanisms (Fingleton 2017).
The aim of this review was to present on the major biological activities of acylated flavonoids, sources, and impact of acylation on their chemical and biological properties. Biological effects discussed as part of this study include antimicrobial, anti-parasitic, anti-inflammatory, anti-complementary, analgesic and anti-nociceptive (Fig. 1). Further, docking simulations have been conducted by Molecular Operating Environment (MOE) employing a validated rigid docking protocol to predict the potential of acylated flavonoids with anti-inflammatory activity to inhibit MMP-2 and -9, where possible accommodation and interaction of such phytochemicals with the active sites amino acids may provide promising insights into their anti-inflammatory mechanism.

A diagrammatic sketch of acylated flavonoids biological effects discussed in text and their main action mechanisms
Experimental section
Scientific literature searches strategies
Our work including overall documented data concerning acylated flavonoids isolated from plants (Fig. 2) with selected bioactivities including antimicrobial (Table 1), anti-parasitic (Table 2), anti-inflammatory (Table 3), anti-nociceptive analgesic (Table 4), and anti-complementary (Table 5) were collected. Literature was summarized from digital databases such as Sci-finder, PubMed, Elsevier, Springer, MDPI, Web of Science and Google Scholar. The data were selected based upon the main biological targets of the current topics from 1970 up to date, with emphasis on more recent years regarding action mechanisms specifically from 2000 till present.



Acylated flavonoids reported in literature with different biological activities
Molecular docking
Docking study was performed employing Molecular Operating Environment (MOE) 2015.10 software. The coordinates of MMP-2 (PDB ID: 1HOV) (Feng et al. 2002) and MMP-9 (PDB ID: 1GKC) (Rowsell et al. 2002), GyrB (PDB ID: 4PRV) (Stanger et al. 2014), mglur1 (PDB ID: 3KS9 (Drobovetsky et al. 2010), and complement component 5a receptor 1(human C5a receptor) (PDB ID: 6C1R (Heng Liu et al. 2018) catalytic domains were retrieved from the protein data bank. The crystal structures were freed from unwanted ligands, and solvents, then prepared employing the default MOE “QuickPrep” module settings. The studied flavonoids MDB database file was built in silico and energy minimized employing Amber10:EHT force field with reaction-field electrostatics (an interior dielectric constant of 1 and an exterior dielectric of 80) using an 8–10 Å cutoff distance. Considering that the active sites of metalloenzymes may be biased to the co-crystalized hydroxamate inhibitors (Esposito et al. 2000), the possible binding sites encompassing the key residues were located by the MOE ‘Site Finder’ feature (El Hawary et al. 2020). Site finder feature was also employed for locating the active site of GyrB. Validation of the docking protocols was performed by redocking the co-crystallized inhibitors in the located binding sites, and reproducing most of the experimental key interactions at acceptable root-mean-square deviation (RMSD). The best binding modes of the studied flavonoids (Figs. 3, 4, 5, 6) were computed using Triangle Matcher placement method and London dG scoring function which generated the top 10 non redundant poses of the lowest energy conformers. Docking results were listed as the S-scores with RMSD values ≤ 2 Å. Two-dimensional (2D) and three-dimensional (3D) representations of the phytoligands interactions were generated and inspected.





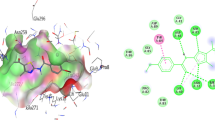
A 3D binding mode of 4 (cyan sticks), B 2D binding mode of 4, C 3D binding mode of 5 (magenta sticks), D 2D binding mode of 5, E 3D binding mode of 6 (blue sticks), F 2D binding mode of 6, G 3D binding mode of 10 (green sticks), and H 2D binding mode of 10, I 3D binding mode of 12 (light pink sticks), J 2D binding mode of 12, K 3D binding mode of 17 (red sticks), L 2D binding mode of 17, M 3D binding mode of 18 (white sticks), N 2D binding mode of 18, O 3D binding mode of 20 (dark orange sticks), P 2D binding mode of 20, Q 3D binding mode of 23 (yellow sticks), and R 2D binding mode of 23, S 3D binding mode of 38 (grey sticks), T 2D binding mode of 38, U 3D binding mode of the crystal structure ligand ADP (orange sticks), and V 2D binding mode of ADP in the active site of E. coli GyrB (PDB ID: 4PRV). The 3D binding modes panels show “dummy atoms” as spheres populating the possible binding sites predicted by MOE Sitefinder feature for docking calculations. Herein, the site 1 comprising 147 amino acid was selected


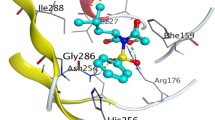
A 3D binding mode of 103, B 2D binding mode of 103 in the active site of MMP-2 (PDB ID: 1HOV), C 3D binding mode of 103, and D 2D binding mode of 103 in the active site of MMP-9 (PDB ID: 1GKC). E 3D binding mode of the MMP-2 co-crystallized ligand (cyan sticks), F 2D binding mode of the cocrystallized ligand in the active site of MMP-2 (PDB ID: 1HOV). G 3D binding mode of the MMP-9 co-crystallized ligand (dark cyan sticks), H 2D binding mode of the cocrystallized ligand in the active site of MMP-9 (PDB ID: 1GKC). The 3D binding modes panels show “dummy atoms” as spheres populating the possible binding sites predicted by MOE Sitefinder feature for docking calculations. Herein, the sites 1,3 and 7 were selected for docking the phytoligand to MMP-2, whereas the site 1 was selected in case of MMP-9

A 3D binding mode of 95 (cyan sticks), B 2D binding mode of 95, C 3D binding mode of 99 (green sticks), and D 2D binding mode of 99, E 3D binding mode of LY341495 (orange sticks), and F 2D binding mode the crystal structure ligand LY341495 in the active site of the metabotropic glutamate receptor mglur1 (PDB ID: 3KS9)


A 3D binding mode of 113 (green sticks), B 2D binding mode of 113, C 3D binding mode of 117 (yellow sticks), D 2D binding mode of 117, E 3D binding mode of 118 (cyan sticks), F 2D binding mode of 118, G 3D binding mode of 123 (white sticks), H 2D binding mode of 123, I 3D binding mode of avacopan (orange sticks), and J 2D binding mode of avacopan in the active site of the human C5a receptor (PDB ID: 6C1R)
Biological activities of acylated flavonoids
While flavonoids are ubiquitous in nature, acylated flavonoids are less common, though with potential biological effects compared to their parent acylated forms. In the present study, acylated flavonoids reported in literature with antimicrobial, anti-parasitic, anti-inflammatory, anti-nociceptive, analgesic and anti-complementary potentialities are discussed in context to their distribution and action mechanisms (Fig. 1 & Tables 1, 2, 3, 4 and 5) as detailed in the next subsection for each biological effect.
Antimicrobial action
Excessive plant production of flavonoids owing to microbial infection (Perumal Samy and Gopalakrishnakone 2010) increases interest about their antimicrobial potential, especially with increasing evidence of antibiotic-resistant organisms i.e., methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin resistant enterococci (VRE) (Cushnie and Lamb 2005). Flavonoids, used solely or synergistically with other antimicrobial agents, can affect pathogen growth through different action mechanisms such as membrane disruption and inhibition of cell envelop synthesis, nucleic acid synthesis, and bacterial toxins (Górniak et al. 2019). The antimicrobial activity of acylated flavonoids would be correlated to its lipophilic nature that facilitate their microbial cell membrane uptake or any other microbial cellular targets. The acylated apigenin glucopyranosides, apigenin 7-O-β-D-(6″-trans-p-coumaroyl)-glucoside (1) and apigenin 7-O-β-D-(6″-trans-p-coumaroyl)-3″-O-acetylglucopyranoside (2, Fig. 2), isolated from Acanthus arboreus, showed activity against Bacillus subtilis with minimum inhibitory concentration (MIC) value of 110.7 and 206.4 µM, respectively (Amer et al. 2004). However, compounds 1 and 2 were reported by Cos et. Al. 2006 inactive since IC50 < 43.3 and 40.3 μM, respectively (Cos et al. 2006).
Biocatalytic acylation of chrysoeriol as chrysoeriol-7-[6‴-O-acetyl-β-D-allosyl-(1 → 2)-6″-O-dodecyl-β-D-glucopyranoside] (3, Fig. 2) isolated from two subspecies of Stachys swainsonii led to the inhibition of two Gram positive (G + ve) bacteria Staphylococcus aureus and Bacillus cereus compared to the non-acylated flavone with MIC 1179.2 and 589.6 µM; respectively (positive control streptomycin with MIC 1.72 µM in both). The results are likely attributed to the increased hydrophobicity achieved by aliphatic acyl chain to improve microbial cell membrane penetration (Mellou et al. 2005). Wang et al. (1989) screened the antimicrobial activity of 3-O-methylated flavonols isolated from Psiadia trineruia leaves as well as their acetylated derivatives against Cladosporium cucumerinum and B. cereus. The results revealed that acetoxylated compounds, 4′-acetyloxy-3,5,7-trimethoxyflavone (4), 3′,4′-diacetyloxy-3,5,7-trimethoxyflavone (5), and 7,4′-diacetyloxy-3,5,8,3′-tetramethoxyflavone (6, Fig. 2) exhibited improved activity against C. cucumerinum fungi with the MIC values of 13.5, 11.7, and 10.9 µM than the non-acetylated ones (respective MIC at 56.2, 46.7, and 43.7 µM), though without affecting B. cereus. Although the biological activities of phenolic compounds depend on the formation of hydrogen bonding with biological targets, certain lipophilicity is required for these activities (Wang et al. 1989). Consequently, increase in antifungal activity may be attributed to the lipophilic nature of acylated compounds which increases their penetration inside fungal cell, where they are further deacylated into the active phenolate ionic form (Wang et al. 1989). Based upon the rules of Cos et al. (2006) the three compounds 4, 5, and 6 were found active, while compound 3 was inactive. The activity of 4, 5, and 6 might be correlated to the acetate groups in the three carbons C-7, -3` and -4` that were absent in 3 (Cos et al. 2006). The galloylated quercetin glycoside, quercetin 3-α-L-arabinopyranoside-2″-gallate (7, Fig. 2), isolated from the leaves of Lasiobema japonica exerted potential inhibitory activity against Escherichia coli with MIC of 170.6 µM (Iwagawa et al. 1990). The antimicrobial effect of the coumaroyl quercetin, quercetin-3-O-(2''-E-p-coumaroyl)-α-rhamnopyranosyl-(1''' → 6'')-β-glucopyranoside (8, Fig. 2), isolated from Leucaeana leucocephala leaves showed inhibitory effect on Salmonella typhimurium, E. coli, and Pseudomonas aeruginosa with a % inhibition at 97, 88, 66%, at concentrations of 19.0, 16.8, and 17.2 µM, respectively compared to ampicillin and clotrimazole, individually as broad spectrum antimicrobial drugs (Mohammed et al. 2015). Compound 8 was more effective as antibacterial agent against S. typhimurium, and E. coli (Cos et al. 2006) than compound 7. That would be attributed for the presence of the trans-p-coumaroyl moiety at C-3 of compound 8.
Two other quercetin 3-O-rhamnosides substituted at 4″ with (E) and (Z)-p-caffeoyl moiety (9 and 10), respectively (Fig. 2) isolated from the root barks of Sophora japonica showed good results against S. aureus and K. pneumonia with MIC of 5.1, 20.5, respectively for compound 9 and 2.4, 20.5 μM, respectively for compound 10, compared to neomycin as standard antibiotic with MIC at 0.64 and 2.54 μM (Si et al. 2016). In contrast, gallate derivative of kaempferol, kaempferol 3-α-rhamnoside-4″-gallate (11, Fig. 2), isolated from the same plant showed potential bactericidal effect against both organisms with MIC values of 1.3 and 10.7 μM, respectively (Si et al. 2016). From these results, acylation of flavonoids with galloyl moiety rather than caffeoyl appears to potentiate the antimicrobial activity against S. aureus and K. pneumonia.
Acylated kaempferol glucopyranosides, stenopalustroside (A-D) (12–15, Fig. 2), were isolated from Stenochlaena palustris leaves and tested for their antibacterial activity compared to chloramphenicol (MIC: 6.2 μM). The compounds with one or two (Z)-p-coumaroyl groups that showed significant activity against B. cereus, Staphylococcus epidermidis, S. aureus, and Micrococcus luteus, with the highest activity for stenopalustroside A (12) at MIC values 5.4, 4.1, 21.6, and 10.8 μM, respectively (Liu et al. 1999). The potential antibiotic potentialities of compounds 12–15 might be attributed to the double bond configuration of p-coumaroyl moiety.
The antibacterial activities of the two acylated kaempferol, tiliroside (16) and platanoside (17, Fig. 2), isolated from Platanus orientails were evaluated against G + ve S. aureus, S. epidermidis and Streptococcus groups B and F and Gram negative (G –ve) Neisseria gonorrhoeae, Haemophilus influenza, E. coli, P. aeruginosa, and K. pneumoniae organisms using disc diffusion method. Compound 16 moderately inhibited all organisms (inhibition zones from 7–20 mm at 0.016–0.51 µM), while 17 inhibited S. aureus (12–15 mm), Streptococcus group B (16–22 mm), E. coli (17–22 mm), K. pneumoniae (10–20 mm), and P. aeruginosa (10–16 mm) tested at 0.016–0.51 µM for all without affecting other organisms in contrast to antibiotics nitrofurantoin (12–26 mm at 1.26 µM), norfioxacin (18–23 mm at 1.56 µM) and trimethoprime (13–30 mm at 1.72 µM) (Mitrokotsa et al. 1993). It could be concluded the presence of more than trans-p-coumaroyl moiety in compound 17 in the structure would rather positively improve its antibiotic activity.
It is worth noting that platanoside (17), showed complete inhibition of human Cytomegalovirus (HCMV) induced antigen at 40 μM without any toxicity for the host cell in contrast to the non acylated compound at 155 μM with 38% inhibition of the viral growth. This increase in antiviral activity is likely due to acyl substituents i.e., p-coumaric acid which prompted to evaluate the activity of the peracetylated derivative (18, Fig. 2) which completely inhibited HCMV- induced antigens at 8.2 μM (IC50 = 3.2 μM), compared to IC50 value of the antiviral drug ganciclovir at 12.9 μM (Mitrokotsa et al. 1993). These results confirmed that acylation improves the antiviral effects in kaempferol acylated glycosides, and has yet to be determined for other flavonol conjugates, and against other viruses to be more conclusive. These documented values of IC50 suggested for the strong antiviral effects of 17 and 18 in which both structures included trans-p-coumaroyl moiety.
Interestingly, Otsuka et al. 2008 isolated two acylated flavonols, kaempferol 3-O-α-L-(2″,4″-di-E-p-coumaroyl)-rhamnoside (19) and kaempferol 3-O-α-L-(2″-Z-p-coumaroyl-4″-E-p-coumaroyl)-rhamnoside (20, Fig. 2), from the leaves of Laurus nobilis. Compounds 19 and 20 were active against not only MRSA with MIC of 1.4–2.8 μM, but also VRE with a value of 5.6–11.2 μM (Otsuka et al. 2008). These results showed that acylation of sugar with p-coumaroyl moiety dramatically increased the activity toward resistant micro-organisms.
Likewise, another three acylated kaempferol rhamnosides, E,Z-platanoside (21), Z,E- platanoside (22), and Z,Z-platanoside (23) besides E,E- platanoside (17, Fig. 2), were isolated from Platanus occidentalis L. and in vitro tested toward (MRSA) (Ibrahim et al. 2009). The data showed that the four compounds exerted bacteriostatic effects with IC50 values of 1.1, 0.97, 0.55, 2.8 μM, respectively, in contrast to the three antibiotics cloxacillin (2.04 μM), and ciprofloxacin (0.30 μM). Considering that (Z)-configured p-coumaroyl conjugates were found more active than those with (E) configuration, change in configuration is thus likely to greatly affect the anti-MRSA activity (Ibrahim et al. 2009). Additionally, Jaswant et al. 2003 isolated kaempferide 3-O-(6″-O-acetyl 4‴-O-α-methylsinapoyl) neohesperidoside (24, Fig. 2) from Aerva tomentosa which revealed a dose dependent bacteriostatic effect against S. aureus and E. coli (Jaswant et al. 2003).
A mixture of four acylated flavonoid glycosides (25–28, Fig. 2), isolated from the roots of Saussurea lappa were evaluated for their antimicrobial activity against nine fungal strains, four Aspergillus species, two Penicillium species, Trichoderma viride, Cladosporium cladosporioides, and Alternaria alternate, and four bacterial organisms, S. aureus, B. subtilis, E. coli, and P. aeruginosa (Rao et al. 2007). The tested samples greatly inhibited all strains with the highest effect recorded for compound 25 with MIC 0.3–10.4 μM as well as minimum fungicidal concentrations (MFCs) in the range of 0.6–12.0 μM compared to miconazole and bifonazole antifungal drugs (0.1–16.1 µM) (Rao et al. 2007). The potential activity of this compound may be attributed to the acetylated acetoxy-dimethylcyclopentenyl moiety linked to C-3′ of scutellarein.
The acylated kaempferol pentaglycoside, kaempferol-3-O-β-D-glucopyransoyl-(1 → 4)-α-L-rhamnopyranosyl-(1 → 6)-β-D-galactopyranoside 7-O-(6′′′′-O-acetyl-β-D-glucopyranosyl-(1 → 3)-[α-L-rhamnopyranosyl-(1 → 2)]-β-D-glucopyranoside (26), was reported to exert significant inhibitory action against Aspergillus niger, A. ochraceus, A. versicolor, A. flavus, Penicillium ochrochloron, P. funiculosum, Trichoderma viride, Cladosporium cladosporioides, and Alternaria alternate with MIC values of 0.6, 0.6, 0.3, 0.6, 1.2, 1.2, 1.2, 12.0, and 6.0 µM respectively (Rao et al. 2007). While the kaempferol hexaglycoside with caffeoyl and acetyl derivative, kaempferol-3-O-β-Dglucopyranosyl(1 → 2)-β-D-(6a′-O-caffeoyl)-glucopyranoside 7-O-(6′′′′-O-acetyl-β-D-glucopyranosyl- (1 → 3)-[α-L-rhamno-pyranosyl-(1 → 2)]-β-D-glucopyranoside (27), exhibited antimicrobial effect with MICs ranging from 2.3—7.8 µM respectively. In the same study, the di-coumaroyl acetyl derivative of kaempferol, kaempferol-3-O-α-L-(2a′,3a′-E-di-p-coumaroyl)-rhamnoside 7-O-(6′′′′-O-acetyl-β-Dglucopyranosyl-(1 → 3)-[α-L-rhamnopyranosyl-(1 → 2)]-β-D-glucopyranoside (28) showed significantly higher inhibition than 26 and 27 against all strains with MICs ranging from 0.24—8.0 µM respectively (Rao et al. 2007).
Interestingly, several acylated and non-acylated 6-hydroxy flavonol glucopyranosides along their aglycones, isolated from Tagetes minuta, were evaluated for their antibacterial effect against three G + ve (M. leteus, S. aureus, and B. subtilis) and three G –ve, P. pikettii, E. coli, and S. setubal, bacteria using agar well-diffusion assay (Shahzadi and Shah 2015). Results revealed that 6-hydroxyquercetin-7-O-β-(6″-galloylglucopyranoside and 6-hydroxyquercetin-7-O-β-glucopyranoside (29 and 30, Fig. 2) inhibited M. luteus strongly than its aglycone with a respective inhibition zone of 19.0 (at 30.1 µM) and 14.2 mm (at 29.6 µM) (Shahzadi and Shah 2015), confirming the role of acylation and glycosylation in the improvement of flavonoid antimicrobial actions.
Tantry et al. (2012) isolated the two acetylated flavonols, elatoside A and B (31 and 32, Fig. 2), from Epimedium elatum and tested for their antimicrobial activities against S. aureus, E. coli, S. typhi, S. dysenteriae, K. pneumonia, and P. aeruginosa (Tantry et al. 2012). Elatoside A (31) showed promising activity against P. aeruginosa with an inhibition zone of 11.0 mm (at 11.9 µM) relative to gentamicin (30.0 mm), while elatoside B (32) inhibited S. aureus, E. coli, and S. typhi of16 mm (at 16.4 µM), 19 (at 19.4 µM), and 20 mm (at 20.4 µM) compared to gentamicin (32.0 mm). The presence of prenyl unit in both compounds is suggested to play a major role in their activities (Tantry et al. 2012). Additionally, 3-acylated pinobanksin derivatives, (butyrate, isobutyrate, isopentanoate, (2-methyl) butyrate, hexanoate, and octanoate (33–38, Fig. 2) isolated from honey bees exhibited activities against Paenibacillus larvae, pathogen of American foulbrood, with a respective IC50 value at (68, 39, 22 and 17 μM) for 33, 35, 37, and 38, respectively in comparison with tylosin as reference drug its IC50 = 0.3 μM (Wilson et al. 2017). The IC50 values of these four compounds showed that 38 is the strongest antimicrobial agent attributed to the alkane substituent in its structure.
These studies obviously reflect the effect of acylation on broadening the strength and spectrum of flavonoids against not only sensitive microbes but rather resistant ones. The aliphatic acyl chains increase lipophilic character and subsequently improve compounds’ leakage through microbial membranes. Additionally, the aromatic moieties increase the ability of acylated compounds for killing microorganisms at low concentration as in case of kaempferol 3-α-rhamnoside-4″-gallate (11). Moreover, the configuration of double bonds in aromatic acyls greatly influenced their antimicrobial action especially for those with (Z)-configured p-coumaroyl and caffeoyl moieties. This mechanistic activity could be observed in several flavonoids such as the improvement of the action of 6-hydroxyquercetin-7-O-β-(6″-galloylglucopyranoside (29) through the acylation of 6-hydroxyquercetin-7-O-β-glucopyranoside (30, Fig. 2).
Docking the potential antimicrobial flavonoids into Gyrase B
Docking of the potential antimicrobial flavonoids was performed by MOE version 2015.10 against the antibacterial target GyrB (PDB ID: 4PRV) catalytic domain. Bacterial DNA gyrase is responsible for controlling the topological state of DNA using the free energy of ATP hydrolysis. It consists of two catalytic subunits; GyrA is responsible for DNA breakage and reunion, while the subunit GyrB contains the ATP-binding site. GyrB is viewed as attractive target for antibacterial agents that can inhibit the ATPase activity of DNA gyrase by blocking the binding of ATP to subunit GyrB (Oblak et al. 2007). Herein, ADP and water molecules were removed, whereas essential hydrogen atoms were added, and the protein was prepared according to the default settings. The active site was located employing the MOE ‘Site Finder’ feature. The studied flavonoids MDB database file was built in silico and energy minimized. After validation through docking back ADP to the active site on GyrB and restoring the co-crystalized ADP orientation and interactions, the validated rigid docking protocol was followed utilizing Triangle Matcher placement method and London dG scoring function. Docking results were listed as the S-scores (Table 1S) and the binding modes were illustrated in Fig. 3. Most of the phyto ligands showed moderate binding affinities compared to the docked reference co-crystallized ADP (Table 1S). Stenopalustroside A (12) was at the top of the list (ΔG = -9.142 kcal/mol) followed by 6, 10, 17, 4, 18, 23, 20, 5 and 38 with ΔG ranging from -8.838 to -7.357 kcal/mol, respectively. 12, 23 and 38 Interacted with the active site Asp73 and Gly102 through hydrogen bonds attaching both the ligands’ phenolic and carbonyl groups. The phenolic ring of 12 displayed an arene-cation interaction with Arg76. Compound 23 showed additional hydrogen bonding to the active site Glu85 and Ser108 with terminal, phenolic and aliphatic hydroxyl groups, respectively. The lowest energy conformers of 4 posed hydrogen bonding interactions with the active site key residue Gly102 through heterocyclic core carbonyl group. Additional π-H bond between the compound active site and Ile78 assisted better fitting in GyrB active site. Similarly, 5 displayed hydrogen bonding interactions with the active site key residue Gly102 through its acetoxy carbonyl oxygen. The most stable conformer of 6 was able to interact with the active site Mg ion through its methoxy group resembling the co-crystalized ligand binding mode. For 10, the docking simulations of its low energy conformer displayed hydrogen bonding interactions with Asp73, Asp105 and Ser108 through both phenolic and aliphatic hydroxyl groups. The phenolic ring also offered π-H bond with the key Gly102. Similarly, the phenolic ring of 20 displayed π-H bond with Gly102, whereas its phenolic hydroxyl group donated hydrogen bond to Glu85. 18 Accepted hydrogen bonds from the active site Gly101 and Arg136, through its acetoxy carbonyl groups. Despite the relatively moderate docking score of 17 (ΔG =—8.707 kcal/mol), none of its conformers interacted to active site residues.
Anti-parasitic action
Parasitic infections globally result in major health problems leading to more than 2 million deaths/ year especially in the developing countries. Different types of protozoa can infect human such as Leishmania (the cause of Leishmaniasis), Trypanosoma (the cause of Trypanosomiasis), Plasmodium (the cause of Malaria), Schistosoma (the cause of Schistosomiasis) and other helminths (the cause of soil-transmitted helminthiases) (Cavalcanti et al. 2017). Flavonoids were reported in several studies to attenuate protozoal, infection especially in case of acylated glycosides (Jucá et al. 2020; Yang et al. 2018).
Tiliroside (16), isolated from Lychnophora markgravii aerial parts was evaluated against axenic amastigote of Leishmania amazonensis. The compound 16 significantly reduced parasite viability to 30.3% at 1.7 μM relative to 2.3% in case of the positive drug control amphotericin B at 0.02 μM (Salvador et al. 2009).
Two catechin gallates (39 and 40, Fig. 2) were isolated from Piptadenia pervillei leaves that showed strong in vitro inhibitory activity against chloroquine-resistant Plasmodium falciparum with IC50 values of 1.2 and 1.0 μM, respectively, relative to the positive control, chloroquine, with IC50 = 0.12 μM (Ramanandraibe et al. 2008). The results revealed that acylation of catechin with gallate dramatically increased itsanti-plasmodial activity with a broad selectivity index (IC50 toxicity on macrophages/IC50 activity on extracellular or intracellular stages of the parasite). The structure activity-relationship investigated by the semisynthetic analogues of compound 39 indicated that ortho-dihydroxy system on the B ring as well as galloyl moiety at the 3 or 5 positions are prerequisites for antiplasmodial activity (Ramanandraibe et al. 2008).
Four acylated kaempferol 3-O-rhamnosides (41–44, Fig. 2) were isolated from Platanus occidentalis, along with four acylated kaempferol 3-O-glucosides (45–48, Fig. 2) from Quercus laceyi. The compounds 41–48 were assessed for their antimalarial activity against drug-sensitive and multidrug-resistant P. falciparum through a SYBR green-based assay (Cai et al. 2016). Both sets of compounds showed potential effects against sensitive parasites with IC50 range (0.5–2.0 μM and 0.6–2.1 μM, respectively), with less potency against drug-resistant parasites with IC50 range (4.0–7.0 μM and 2.1–5 μM, respectively), while compounds 41–44 were found more selective toward the protozoa than others (Cai et al. 2016).
Acetylation of quercetin 3-O-α-L-rhamnopyranoside, isolated from Albizia zygia leaves resulted in two compounds 49 and 50 (Fig. 2) which significantly inhibited P. falciparum strain 3D7 with IC50 of 7.47 and 6.77 μM, respectively, compared to chloroquine with IC50 2.96 × 10–6 mM (Koagne et al. 2020). As the IC50 value of the precursor compound was at 25.1 μM, the appropriate acetylation of flavonoids may result in new lead compounds for the development of potentially active antiplasmodial agents (Koagne et al. 2020).
Additionally, gallocatechin gallate and epigallocatechin gallate (51 and 52, Fig. 2) showed antiprotozoal activity against Trypanosoma brucei rhodesiense (IC50 value of 8.1 and 19.4 μM, respectively) compared to the reference compound, melarsoprol with IC50 0.0065 μM and towards Leishmania donovani (IC50 value of 20.3 and 41.7 μM, respectively) relative to the reference compound, miltefosine (IC50 0.84 μM) (Mukai et al. 2008). Here, the weak activity reported for nonacylated catechins proved the importance of glloyl residues in anti-plasmodial activity (Tasdemir et al. 2006). Determination of the anthelmintic activity of galloylated epigallocatechin (52), isolated from Camellia sinensis, against Caenorhabditis elegans adult worm revealed good inhibitory activity with LC50 of 0.24 μM lower than that of the reference mebendazole at 0.334 μM. Comparing the activity of this compound 52 with the non galloylated epigallocatechin with LC50 1.11 μM confirmed that the galloyl moiety increased the helminthic effect of flavonoids (Mukai et al. 2008).
The acetylated flavonoids, 2″-acetylastragalin, astragalin heptaacetate, 2″-acetylpaenoside, paenoside decaacetate, 2″-acetylpetiolaroside, and petiolaroside decaacetate (53–58, Fig. 2), isolated from Delphinium staphisagria were evaluated in vitro and in vivo for their anti-trypanosomal activities (Marín et al. 2011). All these compounds exhibited good effect with the highest for astragalin hepta acetate (54) and 2″-acetyl astragalin (53) at IC50 values of 0.8 and 6.5 μM and selectivity index (SI) 205.0 and 45.2, respectively relative to benznidazole with IC50 = 15.8 μM and SI 0.9. Ultrastructure analysis as well as metabolism-excretion studies showed changes in acetate and succinate metabolism which may be explained by alterations in mitochondrial enzymatic complex (Marín et al. 2011). Also, the same compounds 53–58 (Fig. 2) were further in vitro evaluated for their anti-leishmanial effect against different forms of L. infantum and L. braziliensis. The petiolaroside-based compounds 57 and 58 showed high potentiality for inhibition of promastigote, axenic amastigote, and intracellular amastigote forms of these parasites. 2″-Acetylpetiolaroside (57) inhibited three forms of L. infantum with IC50 (19.1, 18.4, 32.1 μM, respectively) and L. braziliensis with IC50 29.0, 25.5, 20.1 μM, respectively relative to the reference compound glucantime® “Meglumine Antimoniate” with IC50 values of 18.0, 24.2, 30.0 μM for the first parasite and 25.6, 30.4, 31.1 μM for the latter (Ramírez-Macías et al. 2012). Petiolaroside decaacetate 58 presented IC50 values of 22.8, 21.5, 31.0 μM for forms of L. infantum and values of 32.3, 24.6, 21.0 μM for L. braziliensis (Ramírez-Macías et al. 2012).
Other acetylated derivatives of quercetin, trifolin, and acetyl hyperoside, 59–61 (Fig. 2), isolated from Consolida oliveriana, showed strong effect against promastigote and amastigote (extracellular and intracellular stages, respectively) of Leishmania (V.) peruviana and Leishmania (V.) braziliensis lower than the effective levels of the reference compounds pentostam (11.32 and 9.56 µM) and glucantime® (15.33 and 25.61 µM). Octa-O-acetylhyperoside, 61, was found the most potent inhibitor against both parasites with IC50 values of 7.35 and 6.21 μM, respectively. While compounds 59 (IC50 10.53 and 8.72 μM) and 60 (IC50 11.18 and 46.78 μM) showed lower activities with much higher IC50 values (Ramírez-Macías et al. 2012; Marin et al. 2009). Transmission electronic microscopy (TEM) examination showed an intense vacuolization and disruption of different membranes which may be attributed to inhibition of tubulin polymerization and acetate biosynthesis. These results proved that increase in number of acetyl substituents in flavonoids improved their antiparasitic effect with very low toxicity (Marin et al. 2009).
Several acylated quercetin and kaempferol tetra, tri and diglycosides (62–75, Fig. 2) were reported to show potential anti-trypanosomal and anti-leishmanial against extracellular and intracellular forms of T. cruzi and three Leishmania (L. infantum, L. braziliensis and L. donovani) with high selectivity index (Marín et al. 2017). The compounds 62 and 67 significantly inhibited the epimastigote and amastigote forms of T. cruzi with IC50 value of (16.4, 23.3 μM and 18.3, 21.2 μM, respectively), compared to IC50 of BZN at 15.8 and 23.3 μM. While compounds 64 and 65 potentially inhibited both promastigote and amastigote forms of L. infantum with IC50 (2.5, 4.2 μM and 2.8, 4.7 μM, respectively) relative to glucantime with IC50 at 25.6 and 30.4 μM (Marín et al. 2017). Additionally, compounds 67, 74 inhibited the two forms of L. braziliensis with IC50 at 10.5, 9.7 μM and 11.3, 11.6 μM, respectively, in comparison to glucantime at IC50 values of 18.0 and 24.2 μM. Also, compounds 64, 72 showed higher efficacy against L. donovani in extracellular and intracellular forms with IC50 values of 2.4, 1.8 μM and 4.7, 3.9 μM, respectively, than glucantime with IC50 at 26.6 and 33.3 μM. The mechanistic study using TEM and 1H NMR revealed the presence of severe alterations in the structure and enzymatic systems of different organelles i.e., mitochondria or glycosomes resulting in difference in excretion products of all species (Marín et al. 2017).
A kaempferol derivative, kaempferol-3-O-(2″,6″-di-O-(E)-p-coumaroyl)-β-D-glucopyranoside (76) isolated from Styrax pohlii, showed anti-bilharzial action towards adult worms of Schistosoma mansoni with IC50 value of 35.5 μM compared to the positive control praziquantel with IC50 0.54 μM (Braguine et al. 2012). Ineffectiveness of another kaempferol glucopyranoside acylated at the 2″ and 4″ positions suggested that acylation at C-6″ in the sugar ring may enhance activity of acylated flavonoid derivatives (Braguine et al. 2012), and is a crucial structural motif for activity against Schistosoma. Such effect for other parasites has yet to be tested.
Other galloyl conjugates of naringenin (77 and 78, Fig. 2) isolated from Acacia farnesiana pod showed potent anthelmintic effect toward Haemonchus contortus egg and infective larvae using assays of egg hatching inhibition (EHI) and larval mortality with LC50 = 1.3 and 2.0 μM, respectively for ovicidal and larvicidal activity. Results revealed that the presence of galloyl group was found crucial for its nematocidal activity (Zarza-Albarrán et al. 2020), and whether other acylation groups i.e., acetyl, malonyl etc., would show increment or a decrease in effect has yet to be determined.
As shown from mechanism based assays, the activity of acylated flavonoids against different types of protozoa might be attributed to the ability of these compounds to damage membranes and/or change the mitochondrial enzymatic complex of the parasite leading to growth inhibition. Moreover, studies showed that acylation of flavonoids with galloyl groups enhanced their efficacy towards resistant parasites as reported in case of catechin derivatives 39, 40, 51 and 52. The above data revealed that acylated flavonoids are most active against parasitic infections that might be attributed to the increasing capacities of these compounds to damage the membranes and/or change in mitochondrial enzymatic complex of the parasite and thus inhibit its growth.
Anti-inflammatory action
Inflammation is a complicated defensive response involved in different stages of several diseases to include diabetes, asthma, neurodegenerative diseases, cardiovascular diseases, and cancer. Flavonoids exert their anti-inflammatory effects through several action mechanisms such as suppression of regulatory enzymes and transcription factors, inhibition of eicosanoid generating enzymes, scavenging of free radicals and affecting immune cells. Consequently, the anti-phlogistic effect of acylated flavonoids will be discussed in this section.
The p-coumaroyl flavonol glycoside, kaempferol 3-O-[2″,4″-O-di-(E)-p-coumaroyl] β-D-glucopyranoside (79, Fig. 2) isolated from Quercus ilex leaves, was evaluated for topical anti-inflammatory effect showed significant inhibition of edema formation at 50% inhibitory concentration IC50 0.04 μM greater and more prolonged than that of the reference drug indomethacin at 0.12 μM (Tubaro et al. 1989). This result showed that compound 79 is more effective than that of indomethacin by 3.3 times more effective than indomethacin as an anti-inflammatory agent, which might be ascribed to the potential function of the p-coumaroyl moiety as a substitution.
Amen et al. (2015) isolated three non acylated flavonol 3-O-glycosides, along with kaempferol 3-O-α-L-rhamnopyranosyl-(1 → 6)-O-[β-D-glucopyranosyl-(1 → 2)-4‴-O-acetyl-α-L-rhamnopyranosyl-(1 → 2)]-β-D-galactopyranoside (80) from Tipuana tipu (Benth.) leaves and screened their anti-inflammatory activity using carrageenan-induced rat paw edema model (Amen et al. 2015). Compound 80 showed the highest activity with 46.3% inhibition of edema at 26.5 μM. These results revealed that the length of saccharide residue alongside with the presence of acyl groups greatly influenced anti-phlogistic effect (Amen et al. 2015), which could be attributed to change in bioavailability or binding to the target enzymes (Hollman and Arts 2000).
The potential effect of acetylated isoscutellareins, isoscutellarein-7-O-[6′″-O-acetyl-β-D-allopyranosyl-(1 → 2)]-β-D-glucopyranoside (81), 4′-O-methylisoscutellarein-7-O-[6′″-O-acetyl-β-D-allopyranosyl-(1 → 2)]-β-D-glucopyranoside (82), and isoscutellarein 7-O-[6′″-O-acetyl-β-D-allopyranosyl-(1 → 2)]-6″-O-acetyl-β-D-glucopyranoside (83, Fig. 2) for inhibiting soybean lipo-oxygenase (LOX) showed remarkable anti-inflammatory activity with IC50 values of 310 μM (Charami et al. 2008). The mechanism could be mediated through the antioxidant activity of flavonoids and their capacity for chelating Fe present in the active site of LOX (Charami et al. 2008). Structure activity relationship of the three structures being anti-inflammatory agents could be explained by the role of acetylated sugars in the flavonoid structure with position of acylation found though less influential.
The p-coumaroyl apigenin derivatives, acacetin-7-O-β-D-[α-L-rhamnosyl(1 → 6)]3″-E-p-coumaroyl glucopyranoside (84) and apigenin-7-O-(6″-Z-p-coumaroyl)-β-D-glucopyranoside (85, Fig. 2) were isolated from Chrozophora tinctoria and screened for suppressing inflammatory mediators production such as interleukin-1beta (IL-1β), (Interleukin-6) IL-6, (Tumor necrosis factor alpha) TNF-α, and Prostaglandin E2 (PGE2) in the human peripheral blood mononuclear cells (PBMCs) induced by phytohaemagglutinin (PHA). Compound 84 dose dependently inhibited these mediators, not TNF-α, to nearly normal levels at 100 μM (Abdallah et al. 2015). Herein, the presence of p-coumaroyl moiety versus diglycoside moiety improved inhibition effect in flavonoids.
Van et al. 2020 isolated the acylated kaempferol glycoside named barringoside I (86, Fig. 2) from Barringtonia racemosa and evaluated its suppressive effect on lipopolysaccharide (LPS)-induced nitric oxide (NO) production in Murine leukemia cell line (RAW264.7). Results revealed that it moderately decreased NO levels with an IC50 of 52 μM (Van et al. 2020) likely attributed for the two p-coumaroyl moieties in its structure.
The acylated quercetin, quercetin-3-O-β-D-(2″-acetyl)-glucopyranoside (87, Fig. 2), obtained from Cleome viscosa flowers, was tested against carrageenan-induced edema in rat paw and showed remarkable decrease in swelling of rat paw (Senthamilselvi et al. 2012). This result revealed that 87 exerted anti-phlogistic effect in the second phase of biphasic carrageenan-induced inflammation through suppression of prostaglandin synthesis by 31.6, 51.7, and 45.0% at doses of 98.8, 197.6, 395.3 μM (Senthamilselvi et al. 2012). Efficacy is attributed to the role of acetylaed glucopyranoside substitution in C-3 position of quercetin (Salehi et al. 2020). Likewise, quercetin-3-O-β-D-(2″-galloyl)-glucopyranoside (88, Fig. 2) and its aglycone from Diospyros kaki were tested for their effect on inflammation induced by (LPS) in (RAW 264.7) macrophages (Cho et al. 2016). The compound 88 and quercetin significantly reduced expression of NO, (TNF)-α, (IL)-1β, IL-6 inducible NO synthase (iNOS), Cyclooxygenase (COX)-2, and mitogen-activated protein kinase (MAPKs) by 58 and 15%, respectively at 50 μM on macrophages (Cho et al. 2016). Quercetin was reported as a potential anti-inflammatory agent due to its antioxidant action (Liet al. 2016), with acetylation in compound 88 to potentiate its effect. This result may open the door for the use of the compound 88 as an adjuvant anti-inflammatory agent instead of quercetin which exhibits poor oral bioavailability (Li et al. 2016). Moreover, the inhibition potential of quercetin 3-O-(5″-O-galloyl)-α-L-arabinofuranoside (89, Fig. 2), obtained from Psidium guajava was determined against histamine release from rat mast cells as well as NO production from macrophages RAW 264.7 (Matsuzaki et al. 2010). The results revealed significant suppression at 170.6 μM recording 94.4 and 50.0% inhibition, respectively (Matsuzaki et al. 2010). By comparing 89 to 88 activity and chemical structure, 88 was found more active likely attributed to a glucopyranoside versus arabinofuranoside as well as the position of the galloyl moiety.
Quercetin inhibition effect on COX-2 and iNOS expression with low gastric side effects prompted its modification to produce quercetin 3, 3′, 4′- triacetate (90, Fig. 2) which was evaluated for its antioxidant and anti-inflammatory activities. Results showed that acetylated product 90 inhibited carrageenan-induced paw edema (inhibition: 14–49%) at a dose of 46.7 μM greater than quercetin (inhibition: 2–8%) with lower antioxidant activity (Gusdinar et al. 2011). The expected rational of this enhanced antioxidant-nondependent anti-inflammatory activity is through protection of hydroxyl groups from metabolism and increasing its bioavailability. Accordingly, the acetylation of the flavonoid appeared to improve its inflammation inhibition action.
Furthermore, Gaitén et al. 2011 compared acetylated rutin and quercetin, isolated from Phyllanthus orbicularis towards inflammation inhibition induced by 12-O-tetradecanoyl-13-acetate phrobol (TPA) in mice (Gaitén et al. 2011) compared to non-acetylated forms. The acetylated compounds, quercetin pentaacetate (91) and rutin decaacetate (92, Fig. 2), at doses 20, 40 mM inhibited TPA-induced inflammation 2–3 times stronger than non-acetylated compounds which was consistent with previous studies on quercetin pentaacetate 91 (Chen et al. 2001). Compounds 91 and 92 exhibited anti-inflammatory action than non-acetylated analogues that may be described through the contribution of flavonoids acylation in the improvement of the anti-inflammation effect.
Three acylated chrysoeriol glycosides, ozturkoside A-C (93–95, Fig. 2), were isolated from the acetone extract of Sideritis ozturkii and tested for anti-inflammatory activity using carrageenan-induced hind paw edema (Küpeli et al. 2007a). Küpeli and his co-workers fractionated the S. ozturkii acetone extract over silica gel column and then tested the anti-inflammation of the extract, column fractions and isolated compounds. The acetone extract (17.7–28.3%), along with two column fractions (13.3–24.1% and 15.9–26.3%) showed significant inhibitory action against carrageenan-induced hind paw edema model (Küpeli et al. 2007a). In the same study, ozturkoside A-C 93–95 showed significant inhibition of paw edema with an inhibition percentage (I%) of 13.9–24.6% compared to indomethacin (34.0–40.2%). Analysis of the chemical structures of the three compounds, ozturkoside C 95 was characterized by the presence of p-coumaroyl moiety that may be more effective as anti-inflammatory mediator than caffeoyl moiety in compounds 93 and 94.
Two acylated flavones, isoscutellarein-7-O-[6‴-O-acetyl-β-d-allopyranosyl-(1 → 2)]-β-d-glucopyranoside (96) and isoscutellarein-7-O-[6‴-O-acetyl-β-d-allopyranosyl-(1 → 2)]-6″-O-acetyl-β-D-glucopyranoside were isolated from the aerial parts of Sideritis stricta (97, Fig. 2) (Küpeli et al. 2007b), and assessed for their action in carrageenan induced hind paw edema. Results showed anti-inflammatory activity for a mixture of both compounds than that of each suggestive for antagonistic action. Results reported that 96 and 97 exhibited inhibition of 16.9% at dose 76.7 μM) and 14.6% at dose 72.0 μM respectively, while a 1:1 mixture of these two compounds exhibited inhibition of 24.6% relative to indomethacin (40.2%) at a dose of 28.0 μM. This result is likely mediated through the increased hydroxylation groups in 96 with improved inflammation inhibition than 97 that encompassed one acetate substitution in its sugar. Additionally, three acetylated allose containing isoscutellarein glycosides (98–100, Fig. 2) were isolated from Sideritis brevibracteata and evaluated for their anti-phlogistic effect using different edema models to include TPA-induced mouse ear edema model, carrageenan-induced hind paw edema model, and prostaglandin-induced hind paw edema model (Güvenç et al. 2010). The results showed good anti-inflammatory activities for the three compounds at 149.7, 140.9, and 138.1 μM with inhibition rates ranging from (17.1–29.7%), (13.2–23.4%), and (5.0–9.7%). Compound 100 exhibited the strongest anti-inflammatory action mediated through the presence of acetyl moiety (Güvenç et al. 2010). Moreover, compound 96 and 4′-O-methyl-isoscutellarein-7-O-[6‴-O-acetyl-β-D-allopyranosyl-(1 → 2)]-β-D-glucopyranoside (101) isolated from Stachys subnuda were tested for lipooxygenase (5-LOX) inhibition, with both compounds to demonstrate good inhibitory effect with IC50 values of 63.8 and 1.5 μM (Şen et al. 2019). The free hydroxyl group at C-4′ might be the reason for the improved anti-inflammatory effect of compound 96 than 101.
Additionally, evaluation of the anti-inflammatory activity of 8,3′,4′-trihydroxyflavone-7-O-(6″-O-p-coumaroyl)-β-D-glucopyranoside (102) and (-)-4′-methoxy-7-O-(6″-acetyl)-β-D-glucopyranosyl-8,3′-dihydroxy-flavanone (103, Fig. 2), obtained from Bidens frondosa, revealed significant suppressive activity on NF-κB in 293-NF-κB-luciferase assay stimulated by LPS (Le et al. 2015). Compounds 102 and 103 were found to exert significant dose-dependent anti-inflammation effect against TNF-α, IL-1β, IL-6, and IL-10 at a dose of 1.7, 16.8, and 168.4 μM and 2.0, 19.8, and 197.6 μM, compared to ibuprofen as reference drug at a dose of 0.005, 0.05, and 0.5 μM in RAW 264.7 macrophages (Le et al. 2015). Comparison of the anti-inflammatory activity of (−)(2S)-5,6,7,3′,5′-pentahydroxyflavanone-7-O-β-D-glucopyranoside, isolated from Lippia graveolens var. berlandieri, and its peracetylated derivative (104, Fig. 2) as well as non-glycosylated aglycone, using (TPA)-induced ear edema in mice, revealed activity of the acetylated compound 104 with an inhibition percent of 66.8% at 1.2 μM higher than other non-acetylated compounds (33.73- 64.12%) and comparable to that of indomethacin (78.7%) (Gonzlez-Guëreca et al. 2010). The acetyl groups increased the hydrophobic character that was expected to increase the power of skin penetration in topical applications (González-Guëreca et al. 2010). These results are in accordance with other reports on quercetin tri- and penta- acetate which showed greater inhibition of edema than non-acetylated compounds (Gaitén et al. 2011; Gusdinar et al. 2011).
Additionally, selagin-7-O-(6″-O-acetyl-)-β-D-glucoside (105, Fig. 2), isolated from Cancrinia discoidea, was tested in acute and chronic inflammations rat models (Su et al. 2011). Compound 105 significantly inhibited the paw edema induced by serotonin or carrageenan (acute inflammation) and cotton pellet-induced granuloma (chronic inflammation) at a dose of 38.5 μM with higher activity than indomethacin at 28.0 μM (Su et al. 2011).
Flavonol glucuronides isolated from Polygonum aviculare, including 5 acetylated compounds (106–110, Fig. 2) were evaluated for the inhibition of neutrophil elastase release. Compounds 106, 107, 109, 110 were found to significantly decrease elastase released by neutrophils (P < 0.05) at a concentration of 1 μM, whereas 109 exhibited a similar inhibition action as that of quercetin as a reference control (Granica et al. 2013). The similarity of compounds’ activity might be due to their similar chemical structure, with the acetylation of compound 109 at C-3`` may be the main reason of improved activity than other analogues.
As shown, several mechanisms including scavenging of free radicals, suppression of regulatory enzymes and transcription factors, and inhibition of eicosanoid generating enzymes are responsible for controlling inflammation by acylated flavonoids. Also, increasing of the inflammation inhibition might be ascribed to improved efficacy to inhibit and/or decrease the mediation factors of inflammations such as prostaglandin (PG) and lipoxygenase (LOX) synthesis (Fig. 1). A conclusion of this action, above data showed the positive effect of the acetylation especially the p-coumaroyl moiety followed by galloyl moiety. However, the increased number of hydroxylation sites might be one of the reasons for the improved anti-inflammatory due to increase of electron donation and thus free radical scavenging activity.
Results of molecular docking of anti-inflammatory compounds
In this review, almost all of these biological activities were correlated directly at most to inflammation inhibitory action of these acylated derivatives. Consequently, acylated flavonoids with anti-inflammatory effects were further subjected to docking stimulation (MOE) to identify exactly structural motifs mediating for improved efficacy. Docking of 32 acylated flavonoids reported with anti-inflammatory activities was performed using MOE into MMP-2 (PDB ID: 1HOV) and MMP-9 (PDB ID: 1GKC) catalytic domains. Considering inflammation, MMPs family, especially MMP-2 and -9, have been reported to influence inflammation cascade in a myriad of action mechanisms (Fingleton 2017). The protein structures and the studied flavonoids database were prepared utilizing the default settings. Rigid docking protocol was validated then employed. Docking results were listed as the S-scores (Table 2S) and the binding modes were illustrated in the supplementary data (Fig. 1S and 2S).
Most of the studied phytoligands recorded moderate to promising binding affinities compared to the docked reference co-crystallized MMP-2 inhibitor (Table 2S). Isoscutellarein 7-O-[6‴-Oacetyl-β-D-allopyranosyl-(1 → 2)]-β-D-glucopyranoside (81), hypolaetin 7-O-[6‴-O-acetyl-β-D-allopyranosyl-(1 → 2)]-6″-Oacetyl-β-D-glucopyranoside (99), rutindecaacetate (92), and 8,3′,4′-trihydroxyflavone-7-O-(6″-O-p-coumaroyl)-β-D-glucopyranoside (102) were at the top of the list displaying the best binding affinities (ΔG = -10.311, -10.049, -10.044 and -10.024 kcal/mol, respectively). Slightly lower fitting capacities were observed for isoscutellarein 7-O-[6‴-O-acetyl-β-D-allopyranosyl-(1 → 2)]-β-D-glucopyranoside (96) (ΔG = -9.844 kcal/mol), Quercetin 3-O-β-(2″-galloyl)-glucopyranoside (88) (ΔG = -9.660 kcal/mol), 3′-hydroxy-4′-O-methylisoscutellarein 7-O-[6‴-O-acetyl-β-D-allopyranosyl-(1 → 2)] 6″-O-acetyl-β-D-glucopyranoside (100) (ΔG = -9.148 kcal/mol) and ( −)-4′-methoxy-7-O-(6″-acetyl)-β-D-glucopyranosyl-8,3′-dihydroxyflavanone (103) (ΔG = -9.037 kcal/mol). Moderate fitting ranging from -8.962 to -6.699 kcal/mol was recorded for the rest of the phytoligands.
Obviously, the lowest energy conformer of isoscutellarein 7-O-[6‴-Oacetyl-β-D-allopyranosyl-(1 → 2)]-β-D-glucopyranoside (81) (Table 2S) coordinated the catalytic zinc ion through its sugar part resembling the binding modes of classical zinc-chelating MMP inhibitors. The predicted H-π interactions between the heterocyclic core and the ligand essential Leu83, Thr143 and His120, besides hydrogen bonding with Val117, located the molecule in the hydrophobic S1’channel-like subunit. The binding modes of the other studied phytoligands into MMP-2 active site in comparison to the co-crystallized inhibitor are illustrated in the Supplementary data (Fig. 1S).
On the other hand, binding scores of the docked phytoligands-MMP-9 complexes predicted promising inhibitory potentials (Table 2S), in which several flavonoids were found superior to the co-crystallized inhibitor (ΔG = − 8.297 kcal/mol), as 103 (ΔG = − 9.603 kcal/mol), 101 (ΔG = − 9.508 kcal/mol), 81 (ΔG = − 9.363 kcal/mol), 94 (ΔG = − 9.287 kcal/mol). Furthermore, compounds 89, 92, 96, 97, 98, 109 and 110 were comparable to the reference. Figure 4 showed slightly less fitting with ΔG ranging from − 7.880 to − 7.509 kcal/mol. The remainder of flavonoids were predicted to moderately fit into the MMP-9 active sites (ΔG = − 7.414 to − 6.001 kcal/mol). Chrysoeriol 7-O-[2‴-O-p-coumaroyl-6‴-O-acetyl-β-D-glucopyranosyl-(1 → 2)-β-D-glucopyranoside] (ozturkoside C, 95) recorded the least predicted fitting. The binding modes of all the studied phytoligands into MMP-9 active site are illustrated together with the co-crystallized inhibitor in (Fig. 2S).
The binding mode of the lowest energy conformer of ( −)-4′-methoxy-7-O-(6″-acetyl)-β-D-glucopyranosyl-8,3′-dihydroxyflavanone (103) into the MMP-9 active site (Fig. 4) showed predicted hydrogen bonding interaction between the phenolic groups and Asp182 as well as Glu416. The heterocyclic ring carbonyl oxygen accepted hydrogen from Gly186. In addition, the sugar part participated in 2 hydrogen bonding interactions with Leu188 and the ligand essential residue Glu402 located the molecule in the vicinity of S1 subunit. It is worth mentioning that ( −)-4′-methoxy-7-O-(6″-acetyl)-β-D-glucopyranosyl-8,3′-dihydroxyflavanone (103) was among the predicted promising MMP-2 inhibitors (ΔG = -9.037 kcal/mol), where its binding mode with MMP-2 catalytic domain showed that the phenolic ring posed predicted H-π and π -π interactions between the key amino acid Leu83 and His120, respectively (Fig. 4). The sugar part also displayed predicted H-π interaction with His130, whereas the core phenolic group donated hydrogen bond to Glu121. Thus ( −)-4′-methoxy-7-O-(6″-acetyl)-β-D-glucopyranosyl-8,3′-dihydroxyflavanone (103) could be considered as the most balanced predicted dual MMP-2/MMP-9 inhibitor that warrants for further investigation using in vivo assays or ideally clinical trials.
Anti-nociceptive and analgesic properties
Pain management is considered one of the major health challenges due to several adverse effects resulting from the use of classical therapeutics like non-steroidal anti-inflammatory drugs (NSAIDs), opioids, and corticosteroids. Lately, flavonoids have been paid great attention because not only they have fewer side effects, but also they reduce pain through affecting both central and peripheral nervous systems. Flavonoids have been reported to activate cGMP/PKG/ATP-sensitive K+ channels in neurons, block the central Ca+2 channels, and inhibit the synthesis of prostaglandin (PG), etc. (Ferraz et al. 2020).
The three acylated chrysoeriol glycosides, ozturkoside A-C (93–95, Fig. 2), isolated from Sideritis ozturkii aerial parts as acetone extract were evaluated for their anti-nociceptive effect. The three compounds exhibited strong antinociceptive action with inhibition ratios of 31.0, 28.4 and 25.9% at doses of 60.4, 63.6 and 58.5 μM, respectively compared to acetyl salicylic acid (ASA) (48.9%) at a dose of 55.6 μM (Küpeli et al. 2007a). Their anti-inflammatory effects, ozturkoside C (95) exhibited stronger anti-inflammation and anti-nociceptive than other analogues due to the p-coumaroyl moiety found more crucial for its efficacy than the galloyl moiety (Küpeli et al. 2007a).
Quercetin-3-O-(6″-feruloyl)-β-D-galactopyranoside (111, Fig. 2), isolated from Polygonum viscosum exhibited mild analgesic activity as shown by measuring pain perception time in response to thermal stimuli (Eddy’s hot plate method) (Datta et al. 2004). At 120 min. post administration, 78.1 μM, 111 showed its peak response 19.5 (s), while peak response 494.3 (s) of 17.5 μM of the control drug morphine appeared after 30 min (Datta et al. 2004). Testing the anti-nociceptive effect of acylated chrysoeriol glycosides from Sideritis ozturkii using p-benzoquinone-induced (PBQ)-induced abdominal constriction test revealed significant activity for ozturkoside C (95) with inhibitory rate 25.9% at 58.5 μM with 0/6 ulceration ratio relative to 48.9% inhibition observed by 277.8 μM of (ASA) with a 5/6 ulceration ratio (Küpeli et al. 2007a). Also, acetylated allose containing isoscutellarein glycosides (98–100) isolated from Sideritis brevibracteata exhibited analgesic activity in (PBQ)-induced writhing test at an inhibitory ratio of 37.6, 28.8 and 38.7% at a doses of 149.7 140.9 and 138.1 μM, respectively, compared to 555.6 μM (ASA) that showed an inhibitory ratio of 52.6%. It is noteworthy that these compounds exerted their effects without any ulceration effect, while (ASA) showed 4/6 ulcerogenic activity (Güvenç et al. 2010). So, apart from ulcerogenic effects of aspirin and other NSAIDs and addictive effects of morphine and other opioids, acylated flavonoids can greatly participate in pain management with minimal side effects.
Furthermore, isolated acylated flavonoid, isorhamnetin-3-O-α-L-(6″-E-p-coumaroyl)-rhamnoside (112), from Persicaria glabra exhibited mild analgesic activity using hot plate method (Rajamanickam and Rajamohan 2020). The dose of compound 112 at 328.9 μM and its aglycone isorhamnetin showed pain perception time 10.09 (s) and 12.97 (s), respectively at 90 min in comparison to 7.5 μM morphine sulphate that showed 19.52 (s) perception time at 90 min. However, using the same assay, 662.2 μM of quercetin showed 18.78 (s) perception time after 90 min suggestive that increased efficacy is due to the presence of ortho-dihydroxy system on the B ring and free OH at position 3 of C ring (Rajamanickam and Rajamohan 2020).
The acylated flavonoids specifically and flavonoids in general derived from natural resources were reported to exert potential capabilities for pain treatment and management due to their significant free radical scavenging and anti-inflammatory actions (Fig. 1).
Docking the potential analgesic flavonoids into metabotropic glutamate receptor mglur1
To confirm structural motifs suggested to mediate for analgesic action among acylated flavonoids based on qualitative bioassays, docking against known drug target was attempted. The potential analgesic flavonoids were docked into metabotropic glutamate receptor mglur1 (PDB ID: 3KS9) by MOE version 2015.10. Unessential resides and molecules were removed, and the protein was prepared according to the default settings employing the MOE QuickPrep. protocol. The studied flavonoids MDB database file was built in silico and energy minimized. The proposed docking protocol was validated by docking the co-crystalized antagonist LY341495 into the active site and restoring the experimental interactions. The validated docking protocol utilized Triangle Matcher placement method and London dG scoring function. Docking results were listed as S-scores (Table 3S) and the ligand-active site complexes were illustrated in Fig. 5. The studied flavanoids recorded superior binding affinities compared to the docked reference (Table 3S). Compound 95 recorded the best detected binding affinity (ΔG = − 9.639 kcal/mol). It’s lowest energy conformer posed hydrogen bonding interactions with the active site Asp191 and Ser344 through the phenolic hydroxyl groups. The aliphatic hydroxyls also interacted with the receptor Ser165 and Arg323. Additional hydrogen bond was detected between the acetyl carbonyl oxygen and Lys409 assisting better fitting into the active site. The docking simulations of 99 displayed hydrogen bonding interactions mainly posed by the aliphatic hydroxyl groups to the active site Leu342, Ser344 and Lys409. The phenolic ring offered π-H bond with Ser165 and hydrogen bonding interactions binding the phenolic hydroxyl group to Asp208.
Anti-complementary action
The pathways of the complement activation are typical, including classical and lectin, and alternative with each having its own trigger complex (Noris and Remuzzi 2013). In the typical pathways, exogenous substances would activate it as an antigen–antibody complex. The alternative one is active at low levels in the normal host body, known as the tick over mechanism, allowing the system to be ready for rapid activation (Noris and Remuzzi 2013).
Flavonol acyl glycosides 16, 113–115 (Fig. 2) isolated from Galphimia glauca were evaluated in the classical and alternative complement assays and showed dose dependent ability to reduce red blood cells (RBCs) hemolysis induced by complement system (Müller et al. 1998). Compounds 113 and 114 exhibited the same activity with IC50 of 21 µM. Results revealed that linking of aromatic esters to flavonol glycosides greatly improved their anti-complementary effect (Müller et al. 1998). Compounds 113 and 114 characterized by galloyl substitution on the sugar that could be correlated to their anti-complementary actions. Likewise, tiliroside 16 isolated from Helicteres angustifolia showed good anti-complementary activity against classical and alternative systems with total complement activity (TCA) CH50 of 40.0 and AP50 of 105.0 µM (Yin et al. 2016). These Results proved for the crucial motif of sugar- aromatic side chain skeleton for the strong anti-complementary activity in flavonols (Jung et al. 1998). Whether such pattern exists in other flavonoid subclasses need to be determined to identify more active analogues. These results confirmed the potential role of the trans-p-coumaroyl substitutions as anti-complementary mediators. Additionally, quercetin 3-O-β-D-(2″-galloyl)-glucopyranoside (113) as well as three acylated kaempferol and quercetin glycosides, kaempferol 3-O-ß-D-(6"-p-hydroxybenzoyl)-galactopyranoside (116), quercetin 3-O-ß-D-(6"-feruloyl)-galactopyranoside (117), and quercetin 3-O-ß-D-(2"-galloyl)-glucopyranoside (118, Fig. 2) revealed potential anti-complementary activity towards classical pathway with IC50 ranging from 39.0 to 97.0 µM (Park et al. 1999) compared with rosmarinic acid as positive control (IC50 180.0 µM4). These results demonstrated that acylated kaempferol (116) was more active than acylated quercetin compounds due to C-3` hydroxylation which favor the activity through increasing free radicals scavenging activity.
Moreover, Xi et al. 2012 isolated four flavonoid glycosides substituted with caffeoyl groups (119–122, Fig. 2) from Gnaphalium affine and evaluated their anti-complement activity. Results showed good activity for all compounds with the highest effect observed for luteolin 4′-O-β-D-(6″-E-caffeoyl)-glucopyranoside (119) with IC50 value 75.8 µM (Xi et al. 2012). From the same plant, naringenin-7-O-β-D-(6″-E-caffeoyl)-glucopyranoside (gnaphaffine B (123) showed moderate inhibition towards the classical complement pathway with IC50 value of 778.7 µM (Li et al. 2013), suggestive that flavones were more active than flavanone acylated forms in this assay. Flavonoids were documented to exert significant anti-complementary effects directly related to the increase of oxygenated sites in compounds. Results suggested that acylation of flavonoids improved anti-complementary against the classical and alternative systems.
Docking the potential anti-complementary flavonoids into human C5a receptor
Likewise, the potential anti-complementary flavonoids 113, 117, 118 and 123 were docked into the human C5a receptor (PDB ID: 6C1R) by MOE version 2015.10. After validation of docking, the studied flavonoid was docked into the co-crystalized ligand avacapon. The Docking results (Table 4S) and binding mode (Fig. 6) showed that the recorded binding affinity (ΔG = − 7.106 kcal/mol) and mode of 118 was effective as the docked reference allosteric antagonist avacapon (ΔG = − 8.420 kcal/mol). As shown (Fig. 6) the lowest energy conformer of 118 posed hydrogen bonding interaction with the ligand essential active site Thr303. Hence, it could be postulated that 118 may function as an allosteric antagonist of the human C5a receptor. The other studied flavonoids 113, 117 and 123 displayed slightly lower binding affinities (ΔG ranging from − 6.812 to − 6.223 kcal/mol) with the receptor. Compound 113 interacted through hydrogen bonding with Thr303 by its the glucopyranoside hydroxyl group, whereas its core was able to form two H-π interactions with the receptor’s Leu249. On the other hand, the phenolic hydroxyl groups of 117 were found responsible for the predicted hydrogen bonding interaction with the amino acid residues Thr303 and Cys307. The least energetic conformer of 123 posed its core to interact with Cys307 by two hydrogen bonds. An additional hydrogen bond was predicted involving the receptor’s Trp299 and the phytoligand’s galactopyranoside motif.
In silico assessment of the physicochemical and pharmacokinetic parameters formulating drug-likeness
A brief computational study of the bioavailability and drug-like properties of representative promising acylated flavonoids 11, 16, 54, 79, 95 and 117 was performed using Swiss Absorption, Distribution, Metabolism and Excretion (ADME) software (Daina et al. 2017). The net results were summarized in Table 6. Firstly, the physicochemical parameters formulating Lipiniski rule (Lipinski et al. 1997) for predicting the drug ability were computed for the studied phytochemicals. Herein, optimal lipophilicity (Log P < 5) was predicted for all the studied flavonoids, however all violated the acceptable Lipinski’s limits of molecular weight (< 500), hydrogen bond acceptor (< 10) and hydrogen bond donors (less than 5), except for 54 (only 2 violations). Regarding ADME parameters, the investigated phytochemicals recorded relatively high total polar surface areas with poor intestinal and central nervous system (CNS) absorption. Accordingly, it is recommended to employ various drug delivery techniques for the efficient clinical application of these flavonoids. Finally, they were predicted to be devoid of cytochromes P450 2D6 (CYP2D6) inhibition activities.
Conclusion
Flavonoids represent the most abundant class of plant metabolites in human dietary sources with a myriad of health benefits. Recently, increasing interest has been made towards drug discovery of acylated flavonoids to evade limitations in flavonoids in pharmaceutical dosage forms. The acylation processes not only improved the solubility, stability, and bioavailability of flavonoids, but also improved their affinities toward different cellular targets. Here, 123 acylated flavonoids with specific biological activities as antimicrobial, anti-parasitic, anti-inflammatory, anti-nociceptive, analgesic and anti-complementary were comprehensively reviewed to obtain new insights in highlighting potential sources for these compounds and the structural motifs crucial for improved efficacy. Specifically, the addition of long aliphatic acyl groups appeared to improve flavonoids hydrophobic character and transport across biological membranes. Likewise, aromatic acyl moieties i.e., galloyl enhances the binding affinities to molecular targets by increasing the number of donating and accepting centers. The relative configuration of aromatic moieties i.e., p-coumaroyl (Z/E) significantly affected the interaction with binding pockets in targeted proteins, and has yet to be confirmed in other acyl moieties. Docking simulations of the anti-inflammatory compounds showed moderate to promising binding affinities into MMP-2 and MMP-9 catalytic domains. From all tested compounds, ( −)-4′-Methoxy-7-O-(6″-acetyl)-β-D-glucopyranosyl-8,3′-dihydroxyflavanone 103 exhibited the most balanced predicted dual MMP-2/MMP-9 inhibition. Tiliroside 16 was identified as the most bioactive acylated flavonoid among all reported compounds with multi-bioactive potentialities such as antimicrobial, ant-parasite, and anti-complementary. Hence, the current data shows the potential of acylated flavonoids as a promising source of new lead drugs for pharmaceutical applications, posing their most active ones for future biological testing.
Abbreviations
- 2D:
-
Two-dimensional
- 3D:
-
Three-dimensional
- ADME:
-
Absorption, Distribution, Metabolism and Excretion.
- ASA:
-
Acetyl salicylic acid
- BBB:
-
Blood–brain barrier
- Conc.:
-
Concentration
- COX-2:
-
Cyclooxygenase-2
- G + ve:
-
Gram-positive
- G-ve:
-
Gram-negative
- GyrB:
-
DNA gyrase subunit B of all organisms
- HBA:
-
Number of hydrogen bond acceptors.
- HBD:
-
Number of hydrogen bond donors.
- HIA:
-
Human intestinal absorption.
- Human C5a receptor:
-
Complement component 5a receptor 1
- I%:
-
Inhibition percentage
- IC50 :
-
The half-maximal inhibitory concentration
- IL-1β:
-
Interleukin-1beta
- IL-6:
-
Interleukin 6
- iNOS:
-
Inducible nitric oxide synthase
- IZ:
-
Inhibition Zone
- kcal:
-
Kilocalorie
- LC50 :
-
The lethal concentration 50
- log P:
-
Logarithm of compound partition coefficient between n-octanol and water.
- LOX:
-
Lipooxygenase
- LPS:
-
Lipopolysaccharide
- M.Wt.:
-
Molecular weight
- MAPKs:
-
Mitogen-activated protein kinase
- MFC:
-
Minimum Fungicidal concentration
- mglur1:
-
Metabotropic glutamate receptor 1
- MIC:
-
Minimum inhibitory concentration
- min:
-
Minutes
- mm:
-
Millimeter
- MMP-2:
-
Human matrix metallopeptidase 2 protein
- MMP-9:
-
Human matrix metallopeptidase 9 protein
- MOE:
-
Molecular Operating Environment
- Mol:
-
Mole
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- NO:
-
Nitric oxide
- NROTB:
-
Number of rotatable bonds
- PBMCs:
-
Human peripheral blood mononuclear cells
- PBQ-induced:
-
P-Benzoquinone-induced
- PHA:
-
Phytohaemagglutinin
- RAW 264.7:
-
Murine leukemia cell line
- RBCs:
-
Red blood cells
- RMSD:
-
The root-mean-square deviation
- s:
-
Seconds
- SI:
-
Selectivity index
- TCA:
-
Total complement activity
- TNF-α:
-
Tumor necrosis factor alpha
- VRE:
-
Vancomycin resistant enterococci
- μM:
-
Micro-molar
References
Abdallah HM, Almowallad FM, Esmat A et al (2015) Anti-inflammatory activity of flavonoids from Chrozophora tinctoria. Phytochem Lett 13:74–80. https://doi.org/10.1016/j.phytol.2015.05.008
Abe Y, Tera M, Sasaki N et al (2008) Detection of 1-O-malylglucose: pelargonidin 3-O-glucose-6′′-O-malyltransferase activity in carnation (Dianthus caryophyllus). Biochem Biophys Res Comm 373(4):473–477. https://doi.org/10.1016/j.bbrc.2008.04.153
Amen YM, Marzouk AM, Zaghloul MG, Afifi MS (2015) A new acylated flavonoid tetraglycoside with anti-inflammatory activity from Tipuana tipu leaves. Nat Prod Res 29(6):511–517. https://doi.org/10.1080/14786419.2014.952233
Amer ME, Abou-Shoer MI, Abdel-Kader MS et al (2004) Alkaloids and flavone acyl glycosides from Acanthus arboreus. J Braz Chem Soc 15(2):262–266. https://doi.org/10.1590/S0103-50532004000200016
Ardhaoui M, Falcimaigne A, Ognier S et al (2004) Effect of acyl donor chain length and substitutions pattern on the enzymatic acylation of flavonoids. J Biotechnol 110(3):265–272. https://doi.org/10.1016/j.jbiotec.2004.03.003
Biely P, Cziszárová M, Wong KK, Fernyhough A (2014) Enzymatic acylation of flavonoid glycosides by a carbohydrate esterase of family 16. Biotechnol Lett 36(11):2249–2255. https://doi.org/10.1007/s10529-014-1599-x
Braguine CG, Bertanha CS, Gonçalves UO et al (2012) Schistosomicidal evaluation of flavonoids from two species of Styrax against Schistosoma mansoni adult worms. Pharm Biol 50(7):925–929. https://doi.org/10.3109/13880209.2011.649857
Cai S, Risinger AL, Nair S et al (2016) Identification of compounds with efficacy against malaria parasites from common North American plants. J Nat Prod 79(3):490–498. https://doi.org/10.1021/acs.jnatprod.5b00874
Cavalcanti ÉB, Paulo de EV, Scotti L, Scotti M (2017) Flavonoids from asteraceae as multitarget source of compounds against protozoal diseases. Multi-Scale Approaches Drug Discovery Elsevier, 149–190 https://doi.org/10.1016/B978-0-08-101129-4.00007-2
Charami MT, Lazari D, Karioti A et al (2008) Antioxidant and antiinflammatory activities of Sideritis perfoliata subsp perfoliata. (Lamiaceae). Phytother Res 22(4):450–454. https://doi.org/10.1002/ptr.2333
Chen YC, Shen SC, Lee WR et al (2001) Inhibition of nitric oxide synthase inhibitors and lipopolysaccharide induced inducible NOS and cyclooxygenase-2 gene expressions by rutin, quercetin, and quercetin pentaacetate in RAW 264.7 macrophages. J Cell Biochem 82(4):537–548. https://doi.org/10.1002/jcb.1184
Cho YH, Kim NH, Khan I et al (2016) Anti-inflammatory potential of quercetin-3-O-β-D-(“2”-galloyl)-glucopyranoside and quercetin isolated from Diospyros kaki calyx via Suppression of MAP Signaling Molecules in LPS-induced RAW 264.7 Macrophages. J Food Sci 81(10):2447–2456. https://doi.org/10.1111/1750-3841.13497
Cos P, Vlietinc AJ, Berghe DV, Maes L (2006) Anti-infective potential of natural products: how to develop a stronger in vitro ‘proof-of-concept.’ J Ethnopharmcol 106(3):290–302. https://doi.org/10.1016/j.jep.2006.04.003
Cushnie TT, Lamb AJ (2005) Antimicrobial activity of flavonoids. Inter J Antimicrob Agents 26(5):343–356. https://doi.org/10.1016/j.ijantimicag.2005.09.002
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717–42730. https://doi.org/10.1038/srep42717
Datta B, Datta S, Chowdhury M et al (2004) Analgesic, antiinflammatory and CNS depressant activities of sesquiterpenes and a flavonoid glycoside from Polygonum viscosum. Pharmazie 59(3):222–225. https://doi.org/10.1002/chin.200428184
de Araújo MEM, Franco YE, Messias MC et al (2017) Biocatalytic synthesis of flavonoid esters by lipases and their biological benefits. Planta Med 83(1–02):7–22. https://doi.org/10.1055/s-0042-118883
Drobovetsky E, Khutoreskaya G, Seitova A, Cossar D, Edwards AM, Arrowsmith CH, Bountra C, Weigelt J, Bochkarev A (2010) Ligand binding domain of metabotropic glutamate receptor mGluR5 complexed with glutamate. Unpublished. https://doi.org/10.2210/pdb3lmk/pdb
El Hawary SS, Khattab AR, Marzouk HS et al (2020) In silico identification of SARS-CoV-2 spike (S) protein–ACE2 complex inhibitors from eight Tecoma species and cultivars analyzed by LC-MS. RSC Adv 10(70):43103–43108. https://doi.org/10.1039/D0RA08997D
Esposito EX, Baran K, Kelly K, Madura JD (2000) Docking of sulfonamides to carbonic anhydrase II and IV. J Mol Graph Model 18(3):283–289. https://doi.org/10.1016/s1093-3263(00)00040-1
Feng Y, Likos JJ, Zhu L et al (2002) Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochim Biophys Acta 1598(1–2):10–23. https://doi.org/10.1016/s0167-4838(02)00307-2
Ferraz CR, Carvalho TT, Manchope MF et al (2020) Therapeutic potential of flavonoids in pain and inflammation: mechanisms of action, pre-clinical and clinical data, and pharmaceutical development. Molecules 25(3):762. https://doi.org/10.3390/molecules25030762
Fingleton B (2017) Matrix metalloproteinases as regulators of inflammatory processes. Biochim Biophys Acta 1864(11):2036–2042. https://doi.org/10.1016/j.bbamcr.2017.05.010
Gaitén YIG, Martínez MM, Alarcón AB et al (2011) Anti-inflammatory and antioxidant activity of a methanolic extract of Phyllanthus orbicularis and its derived flavonols. J Essent Oil Res 23(5):50–53. https://doi.org/10.1080/10412905.2011.9700482
González-Guëreca M, Soto-Hernandez M, Martínez-Vázquez M (2010) Isolation of (−)(2S)-5, 6, 7, 3′, 5′-pentahydroxyflavanone-7-O-β-D-glucopyranoside, from Lippia graveolens HBK var berlandieri Schauer, a new anti-inflammatory and cytotoxic flavanone. Nat Prod Res 24(16):1528–1536. https://doi.org/10.1080/14786419.2010.488234
Górniak I, Bartoszewski R, Króliczewski J (2019) Comprehensive review of antimicrobial activities of plant flavonoids. Phytochem Rev 18(1):241–272. https://doi.org/10.1007/s11101-018-9591-z
Granica S, Czerwińska ME, Żyżyńska-Granica B, Kiss AK (2013) Antioxidant and anti-inflammatory flavonol glucuronides from Polygonum aviculare L. Fitoterapia 91:180–188. https://doi.org/10.1016/j.fitote.2013.08.026
Gusdinar T, Herowati R, Kartasasmita R, Adnyana I (2011) Anti-inflammatory and antioxidant activity of quercetin-3, 3’, 4’-triacetate. J Pharmacol Toxicol 6(2):182–188. https://doi.org/10.3923/jpt.2011.182.188
Güvenç A, Okada Y, Akkol EK et al (2010) Investigations of anti-inflammatory, antinociceptive, antioxidant and aldose reductase inhibitory activities of phenolic compounds from Sideritis brevibracteata. Food Chem 118(3):686–692. https://doi.org/10.1016/j.foodchem.2009.05.034
Harborne JB, Williams CA (2000) Advances in flavonoid research since 1992. Phytochemistry 55(6):481–504. https://doi.org/10.1016/s0031-9422(00)00235-1
Hollman PCH, Arts IC (2000) Flavonols, flavones and flavanols–nature, occurrence and dietary burden. J Sci Food Agric 80(7):1081–1093. https://doi.org/10.1002/(SICI)1097-0010(20000515)80:7%3c1081::AID-JSFA566%3e3.0.CO;2-G
Ibrahim MA, Mansoor AA, Gross A et al (2009) Methicillin-resistant Staphylococcus aureus (MRSA)-active metabolites from Platanus occidentalis (American sycamore). J Nat Prod 72(12):2141–2144. https://doi.org/10.1021/np900499q
Iwagawa T, Kawasaki JI, Hase T et al (1990) An acylated flavonol glycoside from Lasiobema japonica. Phytochemistry 29(3):1013–1014. https://doi.org/10.1016/0031-9422(90)80075-R
Jaswant B, Ragunathan V, Sulochana N (2003) A rare flavonol glycoside from Aerva tomentosa Forsk as antimicrobial and hepatoprotective agent. Indian J Chem 42 B: 956–958. https://doi.org/10.1002/chin.200328224
Jucá MM, Cysne Filho FMS, de Almeida JC et al (2020) Flavonoids: biological activities and therapeutic potential. Nat Prod Res 34(5):692–705. https://doi.org/10.1080/14786419.2018.1493588
Jung KY, Oh SR, Park SH et al (1998) Anti-complement activity of tiliroside from the flower buds of Magnolia fargesii. Biol Pharm Bull 21(10):1077–1078. https://doi.org/10.1248/bpb.21.1077
Koagne RR, Annang F, Cautain B et al (2020) Cytotoxycity and antiplasmodial activity of phenolic derivatives from Albizia zygia (DC.) JF Macbr. (Mimosaceae). BMC Complement Altern Med 20(1):1–8. https://doi.org/10.1186/s12906-019-2792-1
Kumar S, Pandey AK (2013) Chemistry and biological activities of flavonoids: an overview. Sci World J. https://doi.org/10.1155/2013/162750
Küpeli E, Şahin FP, Çalış İ et al (2007a) Phenolic compounds of Sideritis ozturkii and their in vivo anti-inflammatory and antinociceptive activities. J Ethnopharmacol 112(2):356–360. https://doi.org/10.1016/j.jep.2007.03.017
Küpeli E, Yeşilada E, Caliş İ, Ezer N (2007b) In vivo anti-inflammatory and antinociceptive activity evaluation of phenolic compounds from Sideritis stricta. Z Naturforsch C 62(7–8):519–525. https://doi.org/10.1515/znc-2007-7-810
Le J, Lu W, Xiong X et al (2015) Anti-inflammatory constituents from Bidens frondosa. Molecules 20(10):18496–18510. https://doi.org/10.3390/molecules201018496
Li J, Huang D, Chen W et al (2013) Two new phenolic glycosides from Gnaphalium affine D Don and Their Anti-Complementary Activity. Molecules 18(7):7751–7760. https://doi.org/10.3390/molecules18077751
Li JY, Han C, Yang J et al (2016) Quercetin, inflammation and immunity. Nutrients 8(3):167. https://doi.org/10.3390/nu8030167
Lipinski CA, Lombardo F et al (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 3:3–25. https://doi.org/10.1016/s0169-409x(00)00129-0
Liu H, Orjala J, Sticher O, Rali T (1999) Acylated flavonol glycosides from leaves of Stenochlaena palustris. J Nat Prod 62(1):70–75. https://doi.org/10.1021/np980179f
Liu H, Kim HR, Deepak RK et al (2018) Orthosteric and allosteric action of the C5a receptor antagonists. Nat Struct Mol Biol 25(6):472–481. https://doi.org/10.1038/s41594-018-0067-z
Maleki SJ, Crespo JF, Cabanillas B (2019) Anti-inflammatory effects of flavonoids. Food Chem 299:125124. https://doi.org/10.1016/j.foodchem.2019.125124
Marin C, Boutaleb-Charki S, Diaz JG et al (2009) Antileishmaniasis activity of flavonoids from Consolida oliveriana. J Nat Prod 72(6):1069–1074. https://doi.org/10.1021/np8008122
Marín C, Ramírez-Macías I, López-Céspedes A et al (2011) In vitro and in vivo trypanocidal activity of flavonoids from Delphinium staphisagria against Chagas disease. J Nat Prod 74(4):744–750. https://doi.org/10.1021/np1008043
Marín C, Díaz JG, Maiques DI et al (2017) Antitrypanosomatid activity of flavonoid glycosides isolated from Delphinium gracile, D. staphisagria, Consolida oliveriana and from Aconitum napellus subsp. Lusitanicum Phytochem Lett 19:196–209. https://doi.org/10.1016/j.phytol.2016.12.010
Matsuzaki K, Ishii R, Kobiyama K, Kitanaka S (2010) New benzophenone and quercetin galloyl glycosides from Psidium guajava L. J Nat Med 64(3):252–256. https://doi.org/10.1007/s11418-010-0400-2
Mellou F, Lazari D, Skaltsa H et al (2005) Biocatalytic preparation of acylated derivatives of flavonoid glycosides enhances their antioxidant and antimicrobial activity. J Biotechnol 116(3):295–304. https://doi.org/10.1016/j.jbiotec.2004.12.002
Mitrokotsa D, Mitaku S, Demetzos C et al (1993) Bioactive compounds from the buds of Platanus orientalis and isolation of a new kaempferol glycoside. Planta Med 59(06):517–520. https://doi.org/10.1055/s-2006-959751
Mohammed RS, El Souda SS, Taie HA et al (2015) Antioxidant, antimicrobial activities of flavonoids glycoside from Leucaena leucocephala leaves. J Appl Pharm Sci 5(06):138–147. https://doi.org/10.7324/JAPS.2015.50623
Mukai D, Matsuda N, Yoshioka Y et al (2008) Potential anthelmintics: polyphenols from the tea plant Camellia sinensis L. are lethally toxic to Caenorhabditis elegans. J Nat Med 62(2):155–159. https://doi.org/10.1007/s11418-007-0201-4
Müller A, Reiter S, Wirth C, Wagner H (1998) Anticomplementary flavonoids from Galphimia glauca. Phytomedicine 5(5):341–345. https://doi.org/10.1016/S0944-7113(98)80015-9
Nagase H, Visse R, Murphy G (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69(3):562–573. https://doi.org/10.1016/j.cardiores.2005.12.002
Noris M, Remuzzi G (2013) Overview of complement activation and regulation. Semin Nephrol 33(6):479–492. https://doi.org/10.1016/j.semnephrol.2013.08.001
Oblak M, Kotnik M, Solmajer T (2007) Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents. Curr Med Chem 14(19):2033–2047. https://doi.org/10.2174/092986707781368414
Otsuka N, Liu M, Shiota S et al (2008) Anti-methicillin resistant Staphylococcus aureus (MRSA) compounds isolated from Laurus nobilis. Biol Pharm Bull 31(9):1794–1797. https://doi.org/10.1248/bpb.31.1794
Page-McCaw A, Ewald AJ, Werb Z (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 8(3):221–233
Park SH, Oh SR, Jung KY et al (1999) Acylated flavonol glycosides with anti-complement activity from Persicaria lapathifolia. Chem Pharm Bull 47(10):1484–1486. https://doi.org/10.1248/cpb.47.1484
Perumal Samy R, Gopalakrishnakone PJ (2010) Therapeutic potential of plants as anti-microbials for drug discovery. J Evid Based Complementary Altern Med 7(3):283–294. https://doi.org/10.1093/ecam/nen036
Rajamanickam M, Rajamohan S (2020) Analgesic activity of flavonoids isolated from Persicaria glabra (wild). Orient Pharm Exp Med 20:71–76. https://doi.org/10.1007/s13596-019-00404-x
Ramanandraibe V, Grellier P, Martin M et al (2008) Antiplasmodial phenolic compounds from Piptadenia pervillei. Planta Med 74(4):417–421. https://doi.org/10.1055/s-2008-1034328
Ramírez-Macías I, Marín C, Díaz JG et al (2012) Leishmanicidal activity of nine novel flavonoids from Delphinium staphisagria. Sci World J. https://doi.org/10.1100/2012/203646
Rao KS, Babu GV, Ramnareddy YV (2007) Acylated flavone glycosides from the roots of Saussurea lappa and their antifungal activity. Molecules 12(3):328–344. https://doi.org/10.3390/12030328
Rowsell S, Hawtin P, Minshull CA et al (2002) Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J Mol Biol 319(1):173–181. https://doi.org/10.1016/S0022-2836(02)00262-0
Roy R, Yang J, Moses MA (2009) Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol 27(31):5287. https://doi.org/10.1200/JCO.2009.23.5556
Salvador MJ, Sartori FT, Sacilotto ACB et al (2009) Bioactivity of flavonoids isolated from Lychnophora markgravii against Leishmania amazonensis amastigotes. Z Naturforsch C 64(7–8):509–512. https://doi.org/10.1515/znc-2009-7-807
Salehi B, Machin L, Monzote L, Sharifi-Rad J, Ezzat SM, Salem MA, Merghany R, El Mahdy N, Sibel Kılıç C, Sytar O, Sharifi-Rad M, Sharopov F, Martins N, Martorell M, Cho WC (2020) Therapeutic potential of quercetin: new insights and perspectives for human health. ACS Omega 5(20):11849–11872
Şen A, Göğer F, Dogan A, Bitis L (2019) Two acylated isoscutellarein glucosides with anti-inflammatory and antioxidant activities isolated from endemic Stachys subnuda Montbret Aucher ex Benth. Acta Chim Slov 66(4):831–838. https://doi.org/10.17344/acsi.2018.4921
Senthamilselvi MM, Kesavan D, Sulochana NJO (2012) An anti-inflammatory and anti-microbial flavone glycoside from flowers of Cleome viscosa. Org Med Chem Lett 2(1):1–5. https://doi.org/10.1186/2191-2858-2-19
Shahzadi I, Shah MM (2015) Acylated flavonol glycosides from Tagetes minuta with antibacterial activity. Front Pharmacol 6:195. https://doi.org/10.3389/fphar.2015.00195
Si CL, An LL, Xie DN et al (2016) New acylated flavonol glycosides with antibacterial activity from root barks of Sophora japoni. Wood Sci Technol 50(3):645–659. https://doi.org/10.1007/s00226-016-0809-1
Stanger FV, Dehio C, Schirmer T (2014) Structure of the N-terminal Gyrase B fragment in complex with ADP⋅ Pi reveals rigid-body motion induced by ATP hydrolysis. PLoS ONE 9(9):e107289. https://doi.org/10.1371/journal.pone.0107289
Sternlicht MD, Werb Z (2001) How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17(1):463–516. https://doi.org/10.1146/annurev.cellbio.17.1.463
Su JY, Li QC, Zhu L (2011) Evaluation of the in vivo anti-inflammatory activity of a flavone glycoside from Cancrinia discoidea (Ledeb.) Poljak. EXCLI J 10:110–116
Sudan S, Rupasinghe HV (2015) Antiproliferative activity of long chain acylated esters of quercetin-3-O-glucoside in hepatocellular carcinoma HepG2 cells. J Exp Biol 240(11):1452–1464. https://doi.org/10.1177/1535370215570828
Tantry MA, Dar JA, Idris A et al (2012) Acylated flavonol glycosides from Epimedium elatum, a plant endemic to the Western Himalayas. Fitoterapia 83(4):665–670. https://doi.org/10.1016/j.fitote.2012.02.003
Tasdemir D, Kaiser M, Brun R et al (2006) Antitrypanosomal and antileishmanial activities of flavonoids and their analogues: in vitro, in vivo, structure-activity relationship, and quantitative structure-activity relationship studies. Antimicrob Agents Chemother 50(4):1352–1364. https://doi.org/10.1128/AAC.50.4.1352-1364.2006
Teles YC, Souza MSR, Souza MD (2018) Sulphated flavonoids: biosynthesis, structures, and biological activities. Molecules 23(2):480. https://doi.org/10.3390/molecules23020480
Tubaro A, Del Negro P, Bianchi P et al (1989) Topical anti-inflammatory activity of a new acylated flavonoid. Agents Actions 26(1–2):229–230. https://doi.org/10.1007/BF02126620
Van QTT, Vien LT, Hanh TTH et al (2020) Acylated flavonoid glycosides from Barringtonia racemosa. Nat Prod Res 34(9):1276–1281. https://doi.org/10.1080/14786419.2018.1560290
Viskupičová J, Ondrejovič M, Šturdík E (2009) The potential and practical applications of acylated flavonoids. Pharmazie 64(6):355–360. https://doi.org/10.1691/ph.2009.9501
Wang Y, Hamburger M, Gueho J, Hostettmann K (1989) Antimicrobial flavonoids from Psiadia trinervia and their methylated and acetylated derivatives. Phytochemistry 28(9):2323–2327. https://doi.org/10.1016/S0031-9422(00)97976-7
Wang S, Alseekh S, Fernie AR, Luo J (2019) The structure and function of major plant metabolite modifications. Mol Plant 12(7):899–919. https://doi.org/10.1016/j.molp.2019.06.001
Whittaker M, Floyd CD, Brown P, Gearing A (1999) Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev 99:2735–2776. https://doi.org/10.1021/cr9804543
Wilson MB, Pawlus AD, Brinkman D et al (2017) 3-Acyl dihydroflavonols from poplar resins collected by honey bees are active against the bee pathogens Paenibacillus larvae and Ascosphaera apis. Phytochemistry 138:83–92. https://doi.org/10.1016/j.phytochem.2017.02.020
Xi Z, Chen W, Wu Z et al (2012) Anti-complementary activity of flavonoids from Gnaphalium affine D. Don Food Chemistry 130(1):165–170. https://doi.org/10.1016/j.foodchem.2011.07.025
Yang B, Liu H, Yang J et al (2018) New insights on bioactivities and biosynthesis of flavonoid glycosides. Trends Food Sci Technol 79:116–124. https://doi.org/10.1016/j.tifs.2018.07.006
Yin X, Lu Y, Cheng ZH, Chen DF (2016) Anti-complementary components of Helicteres angustifolia. Molecules 21(11):1506. https://doi.org/10.3390/molecules21111506
Zarza-Albarrán M, Olmedo-Juárez A, Rojo-Rubio R et al (2020) Galloyl flavonoids from Acacia farnesiana pods possess potent anthelmintic activity against Haemonchus contortus eggs and infective larvae. J Ethnopharmacol 249:112402. https://doi.org/10.1016/j.jep.2019.112402
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Essa, A.F., Teleb, M., El-Kersh, D.M. et al. Natural acylated flavonoids: their chemistry and biological merits in context to molecular docking studies. Phytochem Rev 22, 1469–1508 (2023). https://doi.org/10.1007/s11101-022-09840-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-022-09840-1