
Abstract
Purpose
Development of externally predictive Quantitative Structure–Activity Relationship (QSAR) models for Blood–Brain Barrier (BBB) permeability.
Methods
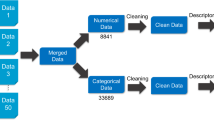
Combinatorial QSAR analysis was carried out for a set of 159 compounds with known BBB permeability data. All six possible combinations of three collections of descriptors derived from two-dimensional representations of molecules as chemical graphs and two QSAR methodologies have been explored. Descriptors were calculated by MolconnZ, MOE, and Dragon software. QSAR methodologies included k-Nearest Neighbors and Support Vector Machine approaches. All models have been rigorously validated using both internal and external validation methods.
Results
The consensus prediction for the external evaluation set afforded high predictive power (R 2 = 0.80 for 10 compounds within the applicability domain after excluding one activity outlier). Classification accuracies for two additional external datasets, including 99 drugs and 267 organic compounds, classified as permeable (BBB+) or non-permeable (BBB−) were 82.5% and 59.0%, respectively. The use of a fairly conservative model applicability domain increased the prediction accuracy to 100% and 83%, respectively (while naturally reducing the dataset coverage to 60% and 43%, respectively). Important descriptors that affect BBB permeability are discussed.
Conclusion
Models developed in these studies can be used to estimate the BBB permeability of drug candidates at early stages of drug development.






Similar content being viewed by others


Abbreviations
- AD:
-
applicability domain
- BBB:
-
blood–brain barrier
- Combi-QSAR:
-
combinatorial QSAR
- kNN:
-
k-nearest neighbors
- MAE:
-
mean absolute error
- NIH:
-
National Institutes of Health
- OECD:
-
Organization for Economic Co-operation and Development
- QSAR:
-
quantitative structure–activity relationship
- SVM:
-
support vector machines
References
P. L. Golden, and G. M. Pollack. Blood–brain barrier efflux transport. J. Pharm. Sci. 92:1739–1753 (2003).
U. Bickel, T. Yoshikawa, and W. M. Pardridge. Delivery of peptides and proteins through the blood–brain barrier. Adv. Drug Deliv. Rev. 46:247–279 (2001).
C. L. Graff, and G. M. Pollack. Drug transport at the blood–brain barrier and the choroid plexus. Curr. Drug Metab. 5:95–108 (2004).
R. C. Young, R. C. Mitchell, T. H. Brown, C. R. Ganellin, R. Griffiths, M. Jones, K. K. Rana, D. Saunders, I. R. Smith, N. E. Sore, and T. J. Wilks. Development of a new physicochemical model for brain penetration and its application to the design of centrally acting H2 receptor histamine antagonists. J. Med. Chem. 31:656–671 (1988).
M. H. Abraham, H. S. Chadha, and R. C. Mitchell. Hydrogen bonding. 33. Factors that influence the distribution of solutes between blood and brain. J. Pharm. Sci. 83:1257–1268 (1994).
M. H. Abraham, H. S. Chadha, and R. C. Mitchell. Hydrogen-bonding. Part 36. Determination of blood brain distribution using octanol–water partition coefficients. Drug Des. Discov. 13:123–131 (1995).
F. Lombardo, J. F. Blake, and W. J. Curatolo. Computation of brain–blood partitioning of organic solutes via free energy calculations. J. Med. Chem. 39:4750–4755 (1996).
G. Subramanian, and D. B. Kitchen. Computational models to predict blood–brain barrier permeation and CNS activity. J. Comput. Aided Mol. Des. 17:643–664 (2003).
D. E. Clark. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood–brain barrier penetration. J. Pharm. Sci. 88:815–821 (1999).
J. M. Luco. Prediction of the brain–blood distribution of a large set of drugs from structurally derived descriptors using partial least-squares (PLS) modeling. J. Chem. Inf. Comput. Sci. 39:396–404 (1999).
M. Feher, E. Sourial, and J. M. Schmidt. A simple model for the prediction of blood–brain partitioning. Int. J. Pharm. 201:239–247 (2000).
J. Kelder, P. D. Grootenhuis, D. M. Bayada, L. P. Delbressine, and J. P. Ploemen. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 16:1514–1519 (1999).
M. E. Brewster, E. Pop, M. J. Huang, and N. Bodor. AM1-based model system for estimation of brain/blood concentration ratios. Int. J. Quantum Chem. 60:51–63 (1996).
U. Norinder, P. Sjoberg, and T. Osterberg. Theoretical calculation and prediction of brain–blood partitioning of organic solutes using MolSurf parametrization and PLS statistics. J. Pharm. Sci. 87:952–959 (1998).
A. Tropsha, P. Gramatica, and V. K. Gombar. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR Models. QSAR Comb. Sci. 22:69–77 (2003).
H. Kubinyi, F. A. Hamprecht, and T. Mietzner. Three-dimensional quantitative similarity–activity relationships (3D QSiAR) from SEAL similarity matrices. J. Med. Chem. 41:2553–2564 (1998).
A. Golbraikh, and A. Tropsha. Beware of q2!. J. Mol. Graph. Model. 20:269–276 (2002).
M. Vracko, V. Bandelj, P. Barbieri, E. Benfenati, Q. Chaudhry, M. Cronin, J. Devillers, A. Gallegos, G. Gini, P. Gramatica, C. Helma, P. Mazzatorta, D. Neagu, T. Netzeva, M. Pavan, G. Patlewicz, M. Randic, I. Tsakovska, and A. Worth. Validation of counter propagation neural network models for predictive toxicology according to the OECD principles: A case study. SAR QSAR Environ. Res. 17:265–284 (2006).
L. P. de Cerqueira, A. Golbraikh, S. Oloff, Y. Xiao, and A. Tropsha. Combinatorial QSAR modeling of P-glycoprotein substrates. J. Chem. Inf. Model. 46:1245–1254 (2006).
A. Kovatcheva, A. Golbraikh, S. Oloff, Y. D. Xiao, W. Zheng, P. Wolschann, G. Buchbauer, and A. Tropsha. Combinatorial QSAR of ambergris fragrance compounds. J. Chem. Inf. Comput. Sci. 44:582–595 (2004).
A. Kovatcheva, A. Golbraikh, S. Oloff, J. Feng, W. Zheng, and A. Tropsha. QSAR modeling of datasets with enantioselective compounds using chirality sensitive molecular descriptors. SAR QSAR Environ. Res. 16:93–102 (2005).
B. Hemmateenejad, R. Miri, M. A. Safarpour, and A. R. Mehdipour. Accurate prediction of the blood–brain partitioning of a large set of solutes using ab initio calculations and genetic neural network modeling. J. Comput. Chem. 27:1125–1135 (2006).
U. Norinder, and M. Haeberlein. Computational approaches to the prediction of the blood–brain distribution. Adv. Drug Deliv. Rev. 54:291–313 (2002).
J. A. Platts, M. H. Abraham, Y. H. Zhao, A. Hersey, L. Ijaz, and D. Butina. Correlation and prediction of a large blood–brain distribution data set—an LFER study. Eur. J. Med. Chem. 36:719–730 (2001).
A. Golbraikh, M. Shen, Z. Xiao, Y. D. Xiao, K. H. Lee, and A. Tropsha. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 17:241–253 (2003).
M. Olah, M. Mracec, L. Ostopovici, R. Rad, A. Bora, N. Hadaruga, I. Olah, M. Banda, Z. Simon, M. Mracec, and Y. I. Oprea. WOMBAT: World of Molecular Bioactivity, in chemoinformatics in drug discovery. Wiley-VCH, New York, 2004.
H. Li, C. W. Yap, C. Y. Ung, Y. Xue, Z. W. Cao, and Y. Z. Chen. Effect of selection of molecular descriptors on the prediction of blood–brain barrier penetrating and nonpenetrating agents by statistical learning methods. J. Chem. Inf. Model. 45:1376–1384 (2005).
L. B. Kier, and L. H. Hall. Molecular connectivity in structure–activity analysis. Wiley, New York, 1986.
L. B. Kier, and L. H. Hall. Molecular connectivity in chemistry and drug research. Academic Press, New York, 1976.
M. Randic. Characterization of molecular branching. J. Am. Chem. Soc. 97:6609–6615 (1975).
L. B. Kier. A Shape index from molecular graphs. Quant. Struct.—Act. Relat. 4:109–116 (1985).
L. B. Kier. Inclusion of symmetry as a shape attribute in Kappa-Index analysis. Quant. Struct.—Act. Relatsh. 6:8–12 (1987).
L. H. Hall, and L. B. Kier. Determination of topological equivalence in molecular graphs from the topological state. Quant. Struct.—Act. Relat. 9:115–131 (1990).
L. H. Hall, B. K. Mohney, and L. B. Kier. The electrotopological state: An atom index for QSAR. Quant. Struct.—Act. Relat. 10:43–51 (1991).
L. H. Hall, B. K. Mohney, and L. B. Kier. The electrotopological state: Structure information at the atomic level for molecular graphs. J. Chem. Inf. Comput. Sci. 31:76–82 (1991).
G. E. Kellogg, L. B. Kier, P. Gaillard, and L. H. Hall. E-state fields: Applications to 3D QSAR. J. Comput. Aided Mol. Des. 10:513–520 (1996).
L. B. Kier, and L. H. Hall. Molecular structure description: The electrotopological state. Academic Press, New York, 1999.
L. B. Kier, and L. H. Hall. A differential molecular connectivity index. Quant. Struct.—Act. Relat. 10:134–140 (1991).
M. Petitjean. Applications of the radius–diameter diagram to the classification of topological and geometrical shapes of chemical compounds. J. Chem. Inf. Comput. Sci. 32:331–337 (1992).
H. J. Wiener. Structural determination of paraffin boiling points. J. Am. Chem. Soc. 69:17–20 (1947).
J. R. Platt. Influence of neighbor bonds on additive bond properties in paraffins. J. Chem. Phys. 15:419–420 (1947).
C. Shannon, and W. Weaver. In mathematical theory of communication. University of Illinois, Urbana, Illinois, 1949.
D. Bonchev, O. Mekenyan, and N. Trinajstic. Isomer discrimination by topological information approach. J. Comput. Chem. 2:127–148 (1981).
A. T. Balaban. Five new topological indices for the branching of tree-like graphs. Theor. Chim. Acta. 53:355–375 (1979).
A. T. Balaban. Highly discriminating distance-based topological index. Chem. Phys. Lett. 89:399–404 (1982).
Talete s.r.l. Dragon. [5.4.2006]. 2007. Milan (Italy).
R. Todeschini, and V. Consonni. Handbook of molecular descriptors. Wiley, Weinheim (Germany), 2000.
W. Zheng, and A. Tropsha. Novel variable selection quantitative structure–property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 40:185–194 (2000).
V. N. Vapnik. In the nature of statistical learning theory. Springer, New York, 2000.
J. R. Votano, M. Parham, L. M. Hall, L. H. Hall, L. B. Kier, S. Oloff, and A. Tropsha. QSAR modeling of human serum protein binding with several modeling techniques utilizing structure-information representation. J. Med. Chem. 49:7169–7181 (2006).
A. Tropsha, and A. Golbraikh. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 13:3494–3504 (2007).
M. Shen, C. Beguin, A. Golbraikh, J. P. Stables, H. Kohn, and A. Tropsha. Application of predictive QSAR models to database mining: Identification and experimental validation of novel anticonvulsant compounds. J. Med. Chem. 47:2356–2364 (2004).
L. Sachs. Applied statistics: A handbook of techniques. Springer, New York, 1984.
K. M. Mahar Doan, J. E. Humphreys, L. O. Webster, S. A. Wring, L. J. Shampine, C. J. Serabjit-Singh, K. K. Adkison, and J. W. Polli. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther. 303:1029–1037 (2002).
G. J. Durant, J. M. Loynes, and H. B. Wright. Potential histamine H2-receptor autagonists. 1. Aminoethylimidayo(1,2-a)pyridines and -imidayo(1,5-a)pyridines. J. Med. Chem. 16:1272–1276 (1973).
P. D. Hansten, and R. H. Levy. Role of P-glycoprotein and organic anion transporting polypeptides in drug absorption and distribution—Focus on H-1-receptor antagonists. Clin. Drug Investig. 21:587–596 (2001).
H. Zhu, A. Tropsha, D. Fourches, A. Varnek, E. Papa, P. Gramatica, T. Oberg, P. Dao, A. Cherkasov, and I. V. Tetko. Combinatorial QSAR modeling of chemical toxicants tested against tetrahymena pyriformis. J. Chem. Inf. Model. in press (2008).
M. Iyer, R. Mishru, Y. Han, and A. J. Hopfinger. Predicting blood–brain barrier partitioning of organic molecules using membrane-interaction QSAR analysis. Pharm. Res. 19:1611–1621 (2002).
W. H. van de, G. Camenisch, G. Folkers, J. R. Chretien, and O. A. Raevsky. Estimation of blood–brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 6:151–165 (1998).
K. Rose, L. H. Hall, and L. B. Kier. Modeling blood–brain barrier partitioning using the electrotopological state. J. Chem. Inf. Comput. Sci. 42:651–666 (2002).
J. R. Votano, M. Parham, L. H. Hall, L. B. Kier, S. Oloff, A. Tropsha, Q. Xie, and W. Tong. Three new consensus QSAR models for the prediction of Ames genotoxicity. Mutagenesis. 19:365–377 (2004).
L. H. Hall, and L. B. Kier. MDL QSAR modeling blood–brain barrier partitioning. http://www.mdl.com/products/pdfs/MDLQSARreprint.pdf. 2002.
T. R. Stouch, and O. Gudmundsson. Progress in understanding the structure–activity relationships of P-glycoprotein. Adv. Drug Deliv. Rev. 54:315–328 (2002).
P. Labute. A widely applicable set of descriptors. J. Mol. Graphics Modell. 18:464–477 (2000).
T. Litman, T. Zeuthen, T. Skovsgaard, and W. D. Stein. Structure–activity relationships of P-glycoprotein interacting drugs: Kinetic characterization of their effects on ATPase activity. Biochim. Biophys. Acta. 1361:159–168 (1997).
T. Suzuki, N. Fukazawa, K. San nohe, W. Sato, O. Yano, and T. Tsuruo. Structure–activity relationship of newly synthesized quinoline derivatives for reversal of multidrug resistance in cancer. J. Med. Chem. 40:2047–2052 (1997).
G. M. Keseru, and L. Molnar. High-throughput prediction of blood–brain partitioning: a thermodynamic approach. J. Chem. Inf. Comput. Sci. 41:120–128 (2001).
T. Salminen, A. Pulli, and J. Taskinen. Relationship between immobilised artificial membrane chromatographic retention and the brain penetration of structurally diverse drugs. J. Pharm. Biomed. Anal. 15:469–477 (1997).
X. L. Ma, C. Chen, and J. Yang. Predictive model of blood–brain barrier penetration of organic compounds. Acta Pharmacol. Sin. 26:500–512 (2005).
A. R. Katritzky, M. Kuanar, S. Slavov, D. A. Dobchev, D. C. Fara, M. Karelson, W. E. Acree Jr., V. P. Solov’ev, and A. Varnek. Correlation of blood–brain penetration using structural descriptors. Bioorg. Med. Chem. 14:4888–4917 (2006).
T. J. Hou, and X. J. Xu. ADME evaluation in drug discovery. 3. Modeling blood–brain barrier partitioning using simple molecular descriptors. J. Chem. Inf. Comput. Sci. 43:2137–2152 (2003).
D. Pan, M. Iyer, J. Liu, Y. Li, and A. J. Hopfinger. Constructing optimum blood brain barrier QSAR models using a combination of 4D-molecular similarity measures and cluster analysis. J. Chem. Inf. Comput. Sci. 44:2083–2098 (2004).
D. A. Winkler, and F. R. Burden. Modelling blood–brain barrier partitioning using Bayesian neural nets. J. Mol. Graph. Model. 22:499–505 (2004).
Acknowledgments
We are grateful to Dr. Scott Oloff for his implementation of the SVM approach that was used in this study. We also thank Dr. J. Grier for his critical comments and his help with editing this manuscript. The studies reported in this paper have been supported by the NIH RoadMap grant GM076059.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhang, L., Zhu, H., Oprea, T.I. et al. QSAR Modeling of the Blood–Brain Barrier Permeability for Diverse Organic Compounds. Pharm Res 25, 1902–1914 (2008). https://doi.org/10.1007/s11095-008-9609-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9609-0